何为功能富集分析?
功能富集分析是将基因或者蛋白列表分成多个部分,即将一堆基因进行分类,而这里的分类标准往往是按照基因的功能来限定的。换句话说,就是把一个基因列表中,具有相似功能的基因放到一起,并和生物学表型关联起来。
何为GO和KEGG?
为了解决将基因按照功能进行分类的问题,科学家们开发了很多基因功能注释数据库,。这其中比较有名的一个就是Gene Ontology(基因本体论,GO)和Kyoto Encyclopedia of Genes and Genomes(京都基因与基因组百科全书,KEGG)。
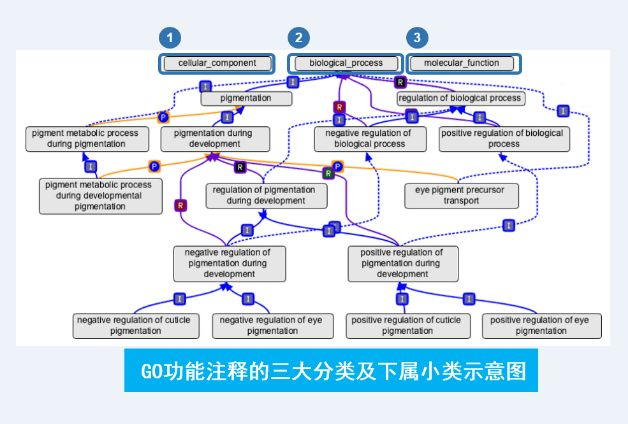
其中,GO是基因本体论联合会建立的一个数据库,旨在建立一个适用于各种物种的、对基因和蛋白功能进行限定和描述的、并能够随着研究不断深入而更新的语义词汇标准。GO注释分为三大类:分子生物学功能(Molecular Function,MF)、生物学过程(Biological Process,BP)和细胞学组分(Cellular Components,CC),通过这三个功能大类,对一个基因的功能进行多方面的限定和描述。
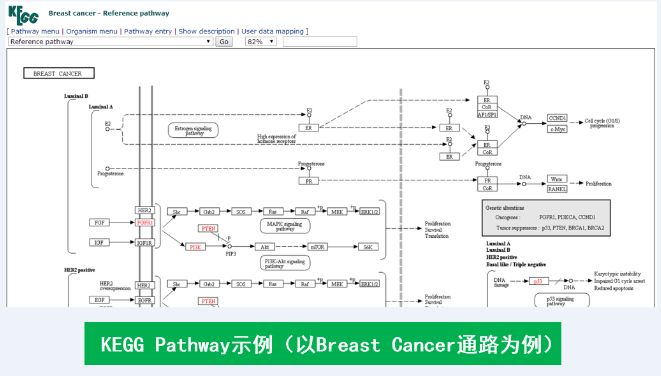
而KEGG,大多数人会将其当做一个基因通路(Pathway)的数据库,其实KEGG的功能远不止于此。KEGG是一个整合了基因组、化学和系统功能信息的综合数据库。KEGG下属4个大类和17个子数据库,而其中有一个数据库叫做 KEGG Pathway,专门存储不同物种中基因通路的信息,也是用的最多的一个,久而久之,KEGG被大家当做一个通路数据库了。
下面两个图展示了GO和KEGG Pathway的面貌。
如何做功能富集分析?
做功能富集分析的算法有很多,能够做功能富集分析的工具也非常多,见下面的列表

Funrich 也可以做功能富集分析
以上的工具中,DAVID最为常用也最为权威。DAVID是由美国Leidos生物医学研究公司的LHRI团队开发的一个在线基因注释及功能富集网站(https://david.ncifcrf.gov/)
使用DAVID做功能富集分析
第一步
打开DAVID官网:https://david.ncifcrf.gov/
点击左侧功能菜单:Functional Annotation
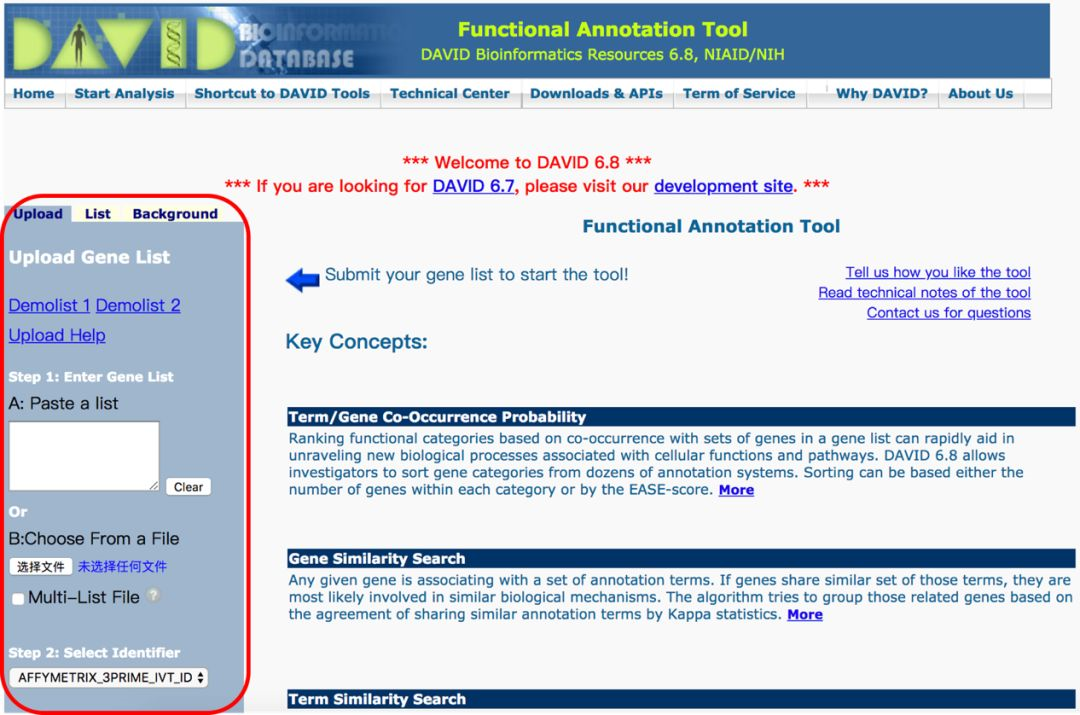
进入到如下的页面中,页面中的红框中就是进行分析所用的主要操作区域。
第二步
进入分析页面后,通过如下三步即可完成分析:
提交基因列表 --> 选定提交列表类型 --> 开始分析
具体操作如下:
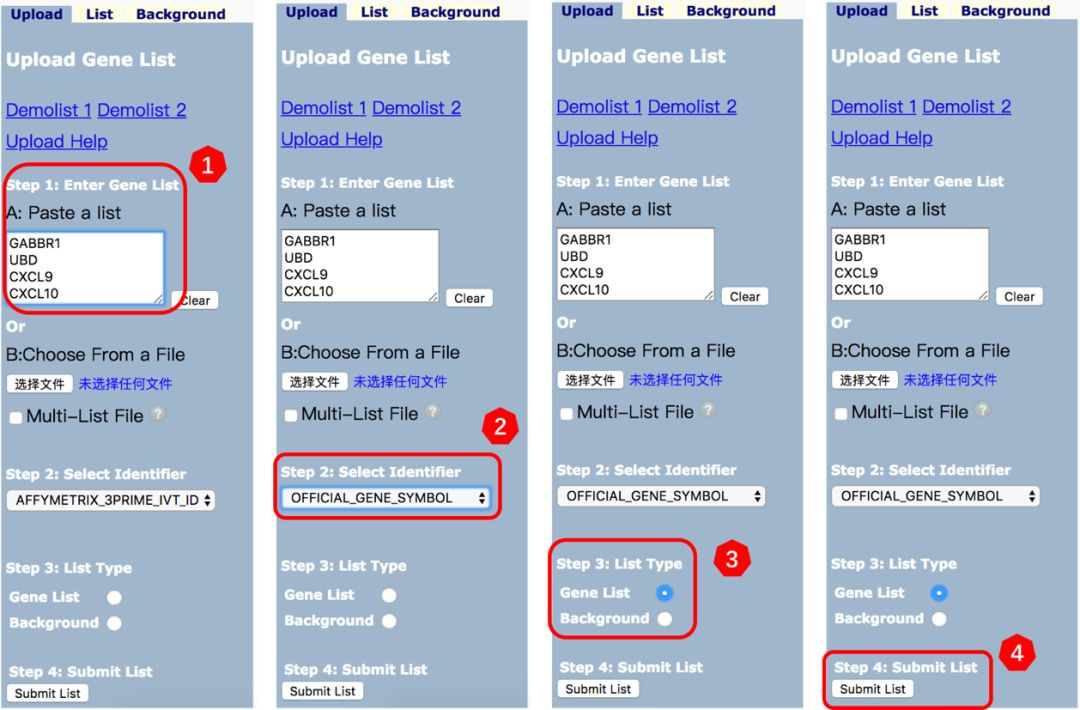
(1) 在 "Enter Gene List" 中上传基因列表,格式是每行一个基因。按照 DAVID 的要求,总的基因个数不得超过 3000 个。
(2) 在 “Select Identifier” 中选择上传的基因类型,因为我们上传的是基因名(Gene Symbol),所以在下拉菜单中选择 “OFFICIAL_GENE_SYMBOL”
(3) 在 “List Type” 中有两个单选框,我们统一选择 “Gene List” 这一项
(4) 点击 “Submit List” 即可
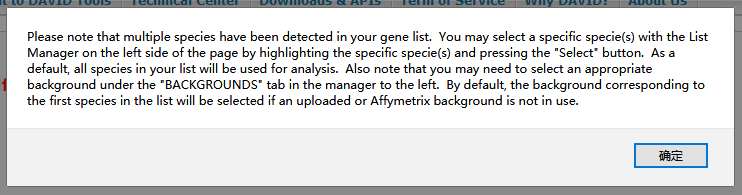
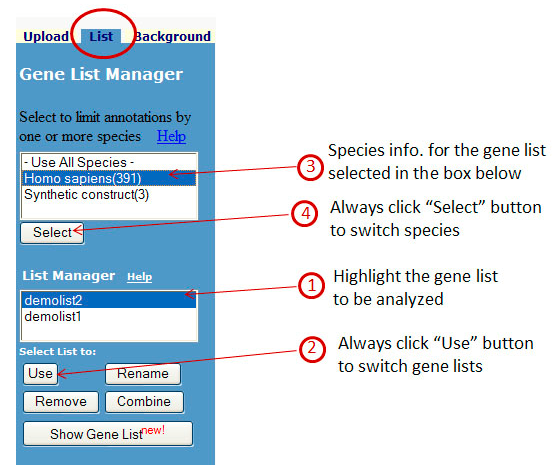
点击 “Submit” 提交基因列表之后,经过几秒钟的等待,如果分析顺利,就会弹出下面一个提示(如下图所示):Please note that multiple species have been detected in your gene list. 这句话的意思就是在我们提交的基因列表中检测到多个物种,需要我们选择相应的物种。怎么选择物种?点击弹出框中的 “确定”,然后在 “List” 中的选择相应物种,这里我们选择 “Homo sapiens”,并点击下方的 “Select Species” 即可。
第三步
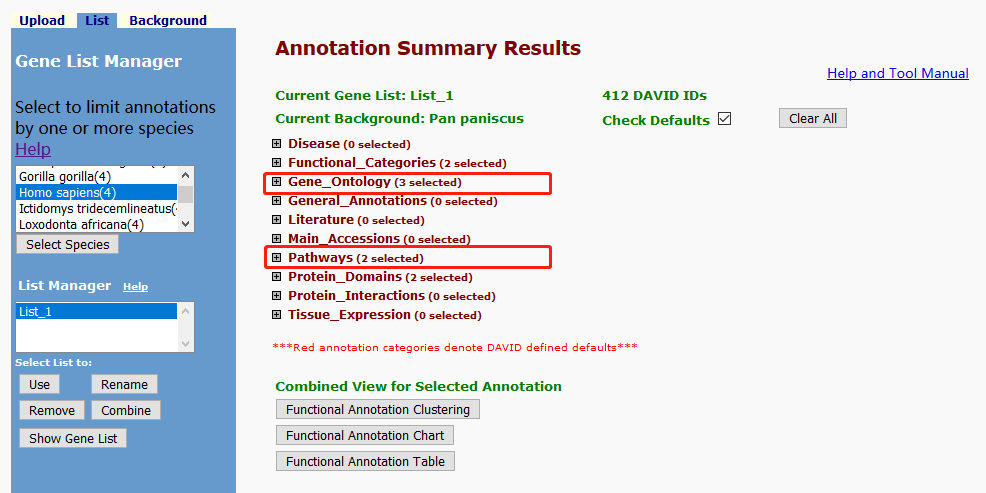
操作完成后,就可以得到如下图所示的分析结果。红框所示折叠框中分别就是GO和KEGG的分析结果。
第四步
做完了分析,我们就来看看如何提取结果,并实现结果的可视化吧。
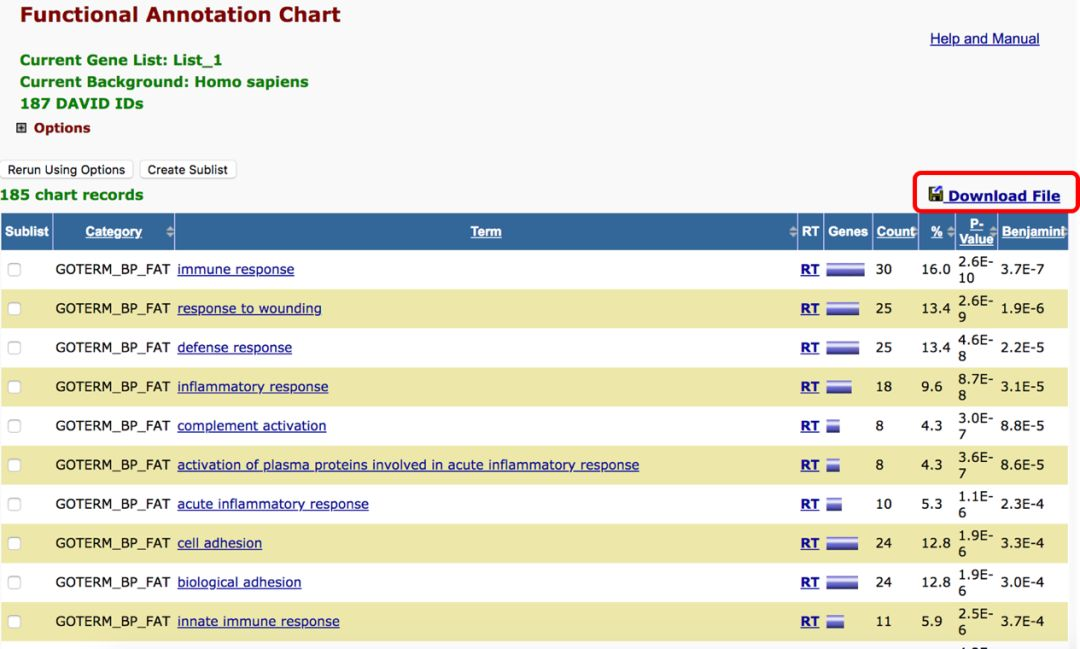
如下图所示,在功能富集分析的结果中有多个折叠栏,其中 Gene_Ontology 这一折叠栏中有有三个栏目:GOTERM_BP_FAT、GOTERM_CC_FAT、GOTERM_MF_FAT 就是我们想要的 GO 功能富集分析结果。而 Pathways 里面有一个 KEGG_PATHWAY 就是我们想要的结果。如何找到 BP、CC、MF 和 KEGG 对应的详细结果呢? 点击每个栏目后面的 “Chart” 即可。

点击 “Chart” 之后,即可出现如下图所示的结果,这里面有几列数据分别是:Category、Term、RT、Genes、Count、%、P-Value 和 Benjamini。这几列中我们比较关心的是:Term(GO语义)、P-Value(P值)、Count(基因数)、%(基因比例)。后面我们要解决的问题是,如何将这些结果下载下来?点击红框中的 Download File 即可。打开一个新的网页,新打开的网页就是分析结果的文本文件,可以下载或者导入到作图软件中进行后续的操作。

第五步 结果导出和可视化
阅读文献时,大家遇到最多的就是柱状图(一般是水平柱状图),柱子的高低与 P-value 相关,柱子越高则越显著。
高级气泡图用来表征富集分析的结果,x轴是 Gene Ratio,对应的就是 DAVID 结果表格中的 % 一列;y轴是富集出来的通路或者 GO Term;点的大小表示 Gene 数;点的颜色最为重要,代表 P值的高低。