转自:https://blog.csdn.net/u012110870/article/details/100171472
1.读取文件
读取h5文件使用函数:
Read10X_h5(atac_peak_file)
读取稀疏矩阵文件
Read10X('路径')
2.求基因活跃矩阵

但是在读取过程中出现了问题,所以尝试下载样例中所用的gtf文件。
但是在Rsutdio中下载超时,则使用https://blog.csdn.net/u011262253/article/details/89363809
wget ftp://ftp.ensembl.org/pub/grch37/release-82/gtf/homo_sapiens/Homo_sapiens.GRCh37.82.gtf.gz
可正常下载。
首先尝试查看小鼠的基因标注文件,https://blog.csdn.net/tanzuozhev/article/details/46536457
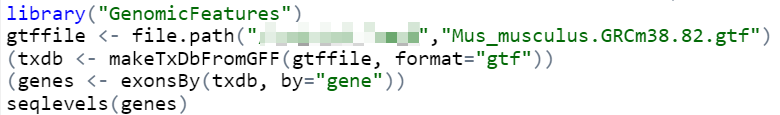

可以看到染色体的名字。那么对应的人类文件中:

这和生物知识也是相符合的,人类有23对染色体,其中22对常染色体,一对性染色体;小鼠有20对染色体,19对常染色体,1对性染色体。
那么范围从c(1:22)修改为c(1:19)即可。也尝试读取一下新下载的小鼠基因注释文件。

![]()
可以发现这里有chr前缀,如果使用它的话,那么seq.levels参数肯定是要改变的。
3.内存溢出问题
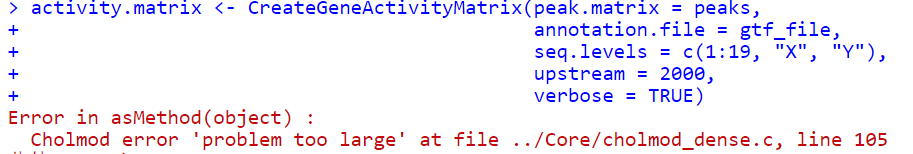
https://stackoverflow.com/questions/62097506/error-in-asmethodobject-cholmod-error-problem-too-large-at-file-core-ch
https://github.com/jumphone/BEER/issues/6,遇到了同样的问题,给出的解决办法是,先存入rds文件中,然后再读取,中间的过程可以释放内存。
尝试使用saveRDS函数:
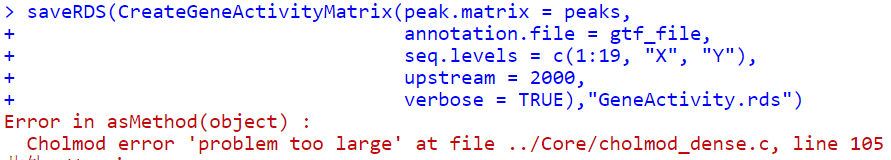
还是同样的错误。尝试重启一下Rstudio
.rs.restartR()
还是不行。
https://github.com/quadbiolab/simspec/issues/4,看到这个终于知道问题出现在哪里了,因为我的这个是稀疏矩阵,它会有转换为as.Matrix的,那么太大,就会出现溢出的情况,那么我先将peaks矩阵转换为rds文件,然后读取。

尝试将其先存入rds,但是它无法转换为matrix文件,实在是太大了,所以最终我选择其子集读取操作。我吐了。