生物纳米分子的“原始数据到完成装配和组装分析”管线与基于序列的基因组FASTA映射

您完成本实验以及示例数据集所需的所有脚本将按照以下说明复制到计算机。您应该按照以下说明,将米色代码块中的文本键入或粘贴到终端中。如果你不习惯命令行,用真实数据练习是最好的学习方式之一。
注意:此管道旨在运行在具有576个内核(48x12核心Intel Xeon CPU),256GB RAM和Linux CentOS 7操作系统的Xeon Phi服务器上。可能需要自定义Irys-scaffolding / KSU_bioinfo_lab / assemble_XeonPhi / rescale_stretch.pl的“自定义RefAligner设置”部分,以在不同的机器上运行BioNano Assembler。也可能需要定制Irys脚手架/ KSU_bioinfo_lab / assemble_XeonPhi / clusterArguments.xml,以便程序集在不同的群集上成功运行。
如果你想要一个基本的linux命令的快速入门,请尝试从软件木工http://software-carpentry.org/v4/shell/index.html这10分钟的课程。
我们将使用从大肠杆菌基因组DNA在BioNano Irys基因组测绘系统上生成的单分子图的BNX文件。我们将准备这些原始分子图,并为他们编写并运行一系列的组件。然后我们将找到最好的装配,并将其用于超级支架,并与大肠杆菌str的片段拷贝进行比较。K-12子 DH10B基因组,并总结了我们的最终装配指标和排列。
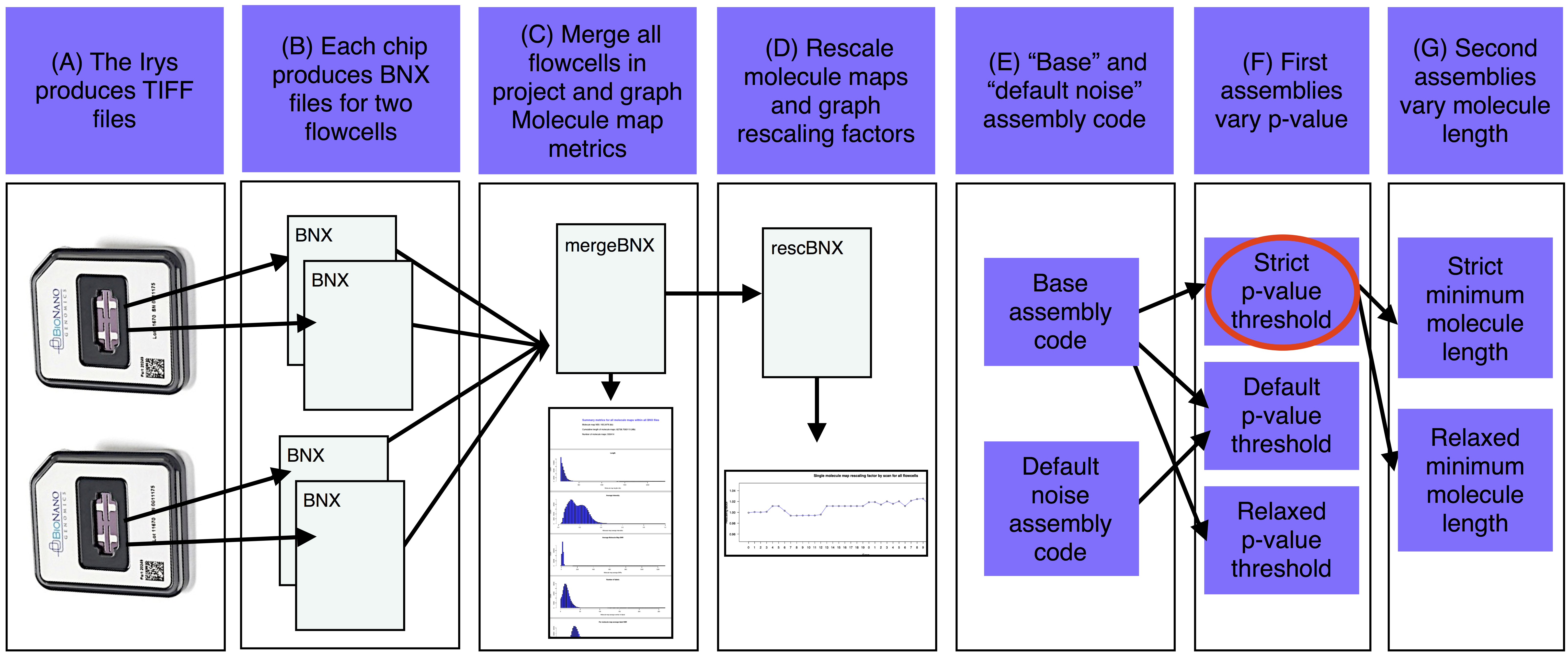
assemble_XeonPhi管道的基本步骤是A)Irys生成转换成分子图的BNX文本文件的TIFF文件。B)每个IrysChip为两个流通池中的每一个生成一个BNX文件。C)每个BNX文件的bnx/子目录-a合并工作目录,汇总和绘制分子图质量度量。D)如果提供了引用,则合并的BNX文件将与序列引用中的计算机映射对齐。拉伸从对准重新缩放,并且每次扫描都会打印重新缩放系数。重新缩放的分子图与参考对齐,估计噪声参数。E)基于估计的基因组大小和噪声参数确定基本汇编代码。F)第一个组件以各种p值阈值运行(至少有一个组件也运行有抑制噪声参数)。G)选择最好的第一个组件(红色椭圆形),并使用各种最小分子长度过滤器生成该组件的版本。
当您通过本实验室,您应该阅读关于软件正在使用通过生成和阅读帮助菜单。
尝试-man标志而不是-help标志更详细的程序描述(您键入q并输入从手动屏幕退出)。
###步骤1:克隆Git存储库
以下工作流程需要安装生物纳米脚本和可执行文件中~/scripts,并~/tools分别目录。按照http://www.bnxinstall.com/training/docs/IrysViewSoftwareInstallationGuide.pdf中的“2.5.1 IrysSolve服务器RefAligner和Assembler”部分中的Linux安装说明进行操作。
完成此操作后,使用以下代码安装KSU自定义软件:
cd ~
git clone https://github.com/i5K-KINBRE-script-share/Irys-scaffolding.git
git clone https://github.com/i5K-KINBRE-script-share/BNGCompare.git
###步骤2:创建具有样本输入数据的项目目录
制作一份工作目录sample_assembly_working_directory。该目录具有大肠杆菌str的片段拷贝。K-12子 DH10B完整的基因组。虽然只有Molecules.bnx文件有任何内容,列出的文件名与IrsyView工作区“Datasets”目录中可以看到的文件名相同。
cp -r ~/Irys-scaffolding/KSU_bioinfo_lab/sample_assembly_working_directory ~
###步骤3:检查刻度密度
阅读本节中的软件:
perl ~/Irys-scaffolding/KSU_bioinfo_lab/map_tools/nick_density.pl -help
perl ~/Irys-scaffolding/KSU_bioinfo_lab/assemble_XeonPhi/third-party/fa2cmap_multi.pl -help
在silico切口基因组FASTA和检查nick密度(为了节省时间,您可以添加--two_enzyme标志跳过除BspQI和BbvCI之外,这两个最常用的酶,如果这两个工作不重新检查所有可能的酶)
perl ~/Irys-scaffolding/KSU_bioinfo_lab/map_tools/nick_density.pl ~/sample_assembly_working_directory/fasta_and_cmap/NC_010473_mock_scaffolds.fa
目的是找到每100 kb具有10至20个切口的酶或酶组合。在这种情况下,结果nick_density.pl表明我们应该使用BspQI酶,每100 kb估计为14.868。
该nick_density.pl脚本创建了可用于计算机图CMAP或标记反应的所有可能的酶的CMAP。您可以使用以下命令查看这些。
ls ~/sample_assembly_working_directory/fasta_and_cmap/cmaps/
如果需要双重切口(例如使用BspQI和BbvCI),因为单酶切痕密度太低,请运行以下命令创建计算机图CMAP。
perl ~/Irys-scaffolding/KSU_bioinfo_lab/assemble_XeonPhi/third-party/fa2cmap_multi.pl -v -i ~/sample_assembly_working_directory/fasta_and_cmap/NC_010473_mock_scaffolds.fna -e BspQI BbvCI
###步骤4:Molecules.bnx从IrysView Dataset子目录获取文件
阅读本节中的软件:
perl ~/Irys-scaffolding/KSU_bioinfo_lab/assemble_XeonPhi/prep_bnxXeonPhi.pl -help
在真实的工作流程中,您可以将Datasets目录从IrysView 移动到装配工作目录并运行prep_bnxXeonPhi.pl。在这种情况下,Datasets目录已经在我们的工作目录中。
perl ~/Irys-scaffolding/KSU_bioinfo_lab/assemble_XeonPhi/prep_bnxXeonPhi.pl -a ~/sample_assembly_working_directory
通过在新bnx子目录中查找Molecule BNX文件来检查它是否有效。您调用的下一个脚本将在bnx程序集中使用组装目录的子目录中的任何BNX文件。
ls ~/sample_assembly_working_directory/bnx
#####请注意,如果您需要直接从Irys创建新的Datasets目录数据:
要创建一个新的Datasets目录,请在您的数据上运行“AutoDetect”。接下来,将所需的流池导入新的IrsyView工作区。导入后,您需要单击工作空间中列出的每个流池才能Molecules.bnx从该RawMolecules.bnx文件生成一个文件。每次点击后,等到RunReportIyrsView显示,然后再移动到下一个流池。最后,将整个Datasets目录移动到您的linux机器和与本实验室相同的工作流程来分析您自己的数据。
###步骤5:准备分子图(即Molecules.bnx文件中的映射)并编写汇编脚本
阅读本节中的软件:
perl ~/Irys-scaffolding/KSU_bioinfo_lab/assemble_XeonPhi/AssembleIrysXeonPhi.pl -help
运行AssembleIrysXeonPhi.pl以生成您的分子图的摘要指标MapStatsHistograms.pdf,以及BNX文件中每次扫描的重新缩放因子bnx_rescaling_factors.pdf。运行AssembleIrysXeonPhi.pl还将输出一个名为的程序集脚本assembly_commands.sh,其中包含具有各种参数的程序集的命令。每组参数都有自己的脚本创建的输出子目录。
perl ~/Irys-scaffolding/KSU_bioinfo_lab/assemble_XeonPhi/AssembleIrysXeonPhi.pl -a ~/sample_assembly_working_directory -g 5 -p Esch_coli_1_2015_000 -r ~/sample_assembly_working_directory/fasta_and_cmap/cmaps/NC_010473_mock_scaffolds_BspQI.cmap
浏览此脚本在~/sample_assembly_working_directory/目录中的输出。
该~/sample_assembly_working_directory/Esch_coli_1_2015_000/MapStatsHistograms.pdf文件包含有关分子图> 100 kb的信息。该信息包括分子图N50和累积长度,图数,分子图信噪比(SNR),分子图强度,每分子图的平均标记SNR和每分子图的平均标记强度。
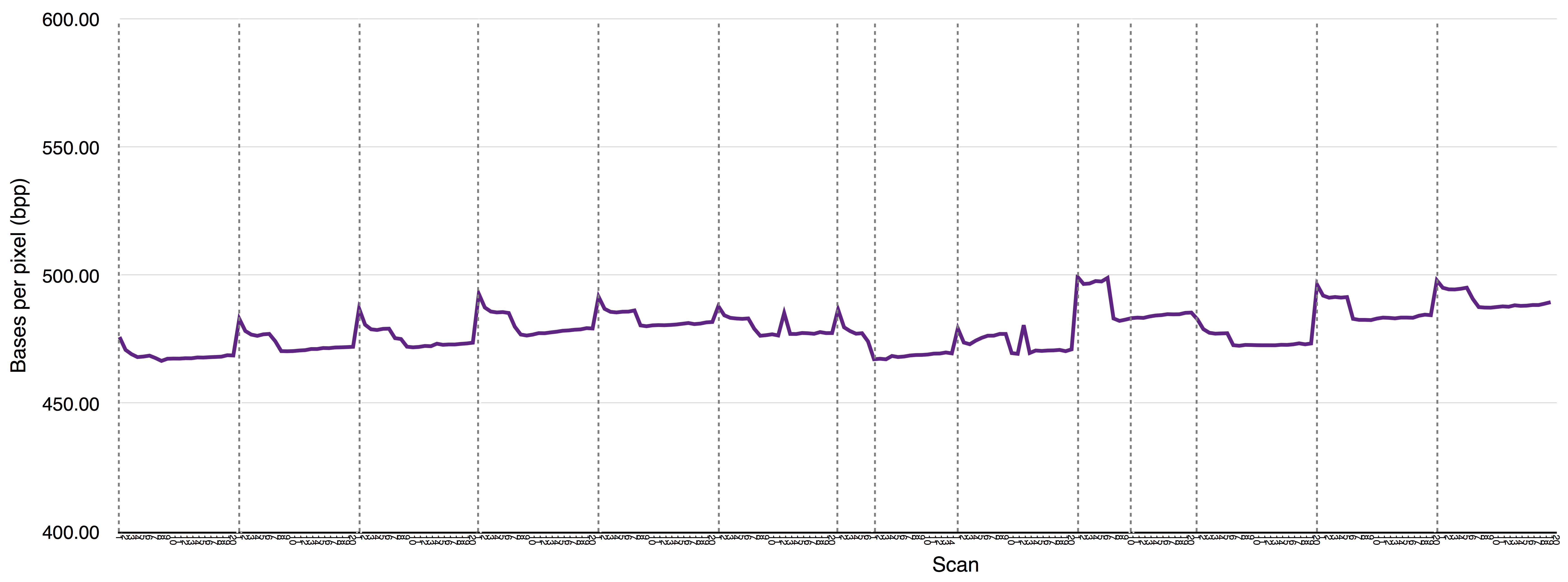
该~/sample_assembly_working_directory/Esch_coli_1_2015_000/bnx_rescaling_factors.pdf文件显示BNX文件的每次扫描的重新调整因子。此输出会因机器和IrysChip版本而异。它也受到Irys上运行的样品与用于组装基于序列的参考样品之间的标记基序相似度的影响。在您的机器上,您可能会注意到高质量BNX文件的可预测模式。一个这样的模式的示例如下所示:
汇编脚本~/sample_assembly_working_directory/assembly_commands.sh是写入除了四个程序集命令之外的所有注释掉的。如果在运行此命令后,没有创建令人满意的程序集,则具有更高和/或更低最小分子图长度以及最佳组合p值阈值的卸载组件。还要注释掉已经运行并保存脚本的程序集。重新运行改变的脚本,看看新参数是否改进了程序集。
###步骤6:运行汇编脚本
阅读本节中的软件:
python2 ~/scripts/pipelineCL.py -help
使用以下命令启动您的前四个程序集:
nohup bash ~/sample_assembly_working_directory/assembly_commands.sh &> ~/sample_assembly_working_directory/assembly_commands_out.txt
###步骤7:评估你的程序集
阅读本节中的软件:
perl ~/Irys-scaffolding/KSU_bioinfo_lab/assemble_XeonPhi/assembly_qcXeonPhi.pl -help
检查您的程序集的质量assembly_qcXeonPhi.pl。
perl ~/Irys-scaffolding/KSU_bioinfo_lab/assemble_XeonPhi/assembly_qcXeonPhi.pl -a ~/sample_assembly_working_directory -g 5 -p Esch_coli_1_2015_000
最终目标通常是产生可用于指导基于序列的单倍体参考基因组装配的共有基因组图谱。虽然单分子图可用于重建单倍型,但基因组组装涉及将多态性任意塌陷到共有参考基因组中。因此,理想的共有基因组图谱的累积长度应等于估计的单倍体基因组长度。另外,100%的共识基因组图将非冗余地对齐到计算机图中的100%。在实践中,最佳的BioNano装配是基于与计算机图中参考的估计的单倍体基因组长度的相似性和最小对准冗余度来选择的。“对齐覆盖宽度”和“总对齐长度”之间的差异越大,对齐冗余越大。
例如,在下图中,Strict-T装配是最好的装配体,因为它的累积尺寸接近200 Mb,估计的基因组大小,以及非冗余对齐长度或“宽度对齐覆盖“和”总对齐长度“。
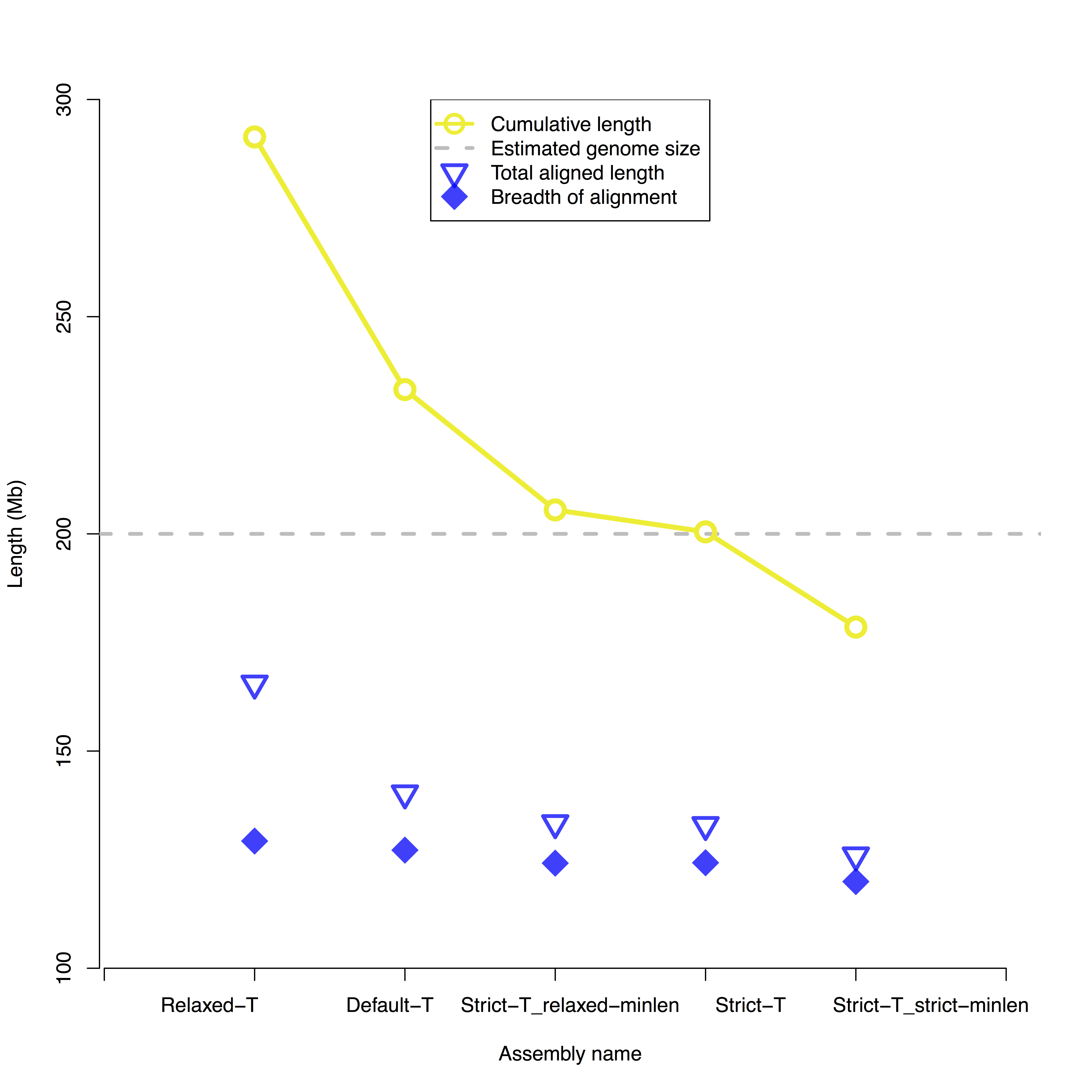
看看~/sample_assembly_working_directory/Assembly_parameter_tests.pdf文件看看这个程序集的结果。
该文件~/sample_assembly_working_directory/Assembly_parameter_tests.csv有关每个程序集的其他详细信息,如果在查看后没有明确的最佳程序集可以使用~/sample_assembly_working_directory/Assembly_parameter_tests.pdf。
###步骤8:将您的最佳组合与电子地图中的参考进行比较
阅读本节中的软件:
perl ~/Irys-scaffolding/KSU_bioinfo_lab/stitch/sewing_machine.pl -help
sewing_machine.pl是编译汇编指标的脚本,并在所有可能的目录中执行“最佳”程序集的拼接:“strict_t”,“default_t”,“relaxed_t”等
针迹过滤器通过置信度对齐XMAP文件,并且对齐的最大潜在长度的百分比。置信度的第一个设置和对齐的全部潜在长度的最小百分比应该被设置为包括在查看原始XMAP之后研究人员决定代表高质量对齐的范围。由于标签密度低或基于短序列的支架长度,某些比对低于最佳置信度。第二组滤波器应具有用户定义的较低最小置信度分数,但是为了捕获这些对准,该比对的最大潜在长度的百分比高得多。应在IrysView中检查结果过滤的XMAP,以查看对齐方式与用户手动选择的一致。每次跑步时,都会找到最好的超级脚手架对齐。run_compare.pl 直到所有超级脚手架都被发现。
我们将从置信度分数(--f_con和--s_con)的可能的对齐阈值(--f_algn和--s_algn)的百分比开始默认过滤参数。一般来说,我们从默认参数开始,然后测试或多或少的严格选项,如果我们的第一个结果不令人满意
perl ~/Irys-scaffolding/KSU_bioinfo_lab/stitch/sewing_machine.pl -b ~/sample_assembly_working_directory/strict_t_150 -p Esch_coli_1_2015_000 -e BspQI -f ~/sample_assembly_working_directory/fasta_and_cmap/NC_010473_mock_scaffolds.fna -r ~/sample_assembly_working_directory/fasta_and_cmap/cmaps/NC_010473_mock_scaffolds_BspQI.cmap
###步骤9:选择最佳对齐参数,并总结您的结果
阅读本节中的软件:
perl ~/Irys-scaffolding/KSU_bioinfo_lab/assemble_XeonPhi/write_report.pl -help
打开~/sample_assembly_working_directory/NC_010473_mock_scaffolds_BNGCompare.csv文件找到最佳对齐参数。像选择最好的组件一样,您想要找到平衡灵敏度(即长整体对齐长度)的结果,而不会过度增加对齐冗余。
perl ~/Irys-scaffolding/KSU_bioinfo_lab/assemble_XeonPhi/write_report.pl -b ~/sample_assembly_working_directory/strict_t_150 -p Esch_coli_1_2015_000 -e BspQI -f ~/sample_assembly_working_directory/fasta_and_cmap/NC_010473_mock_scaffolds.fna -r ~/sample_assembly_working_directory/fasta_and_cmap/cmaps/NC_010473_mock_scaffolds_BspQI.cmap --alignment_parameters default_alignment
###第10步:在IrysView中探索你的结果
读取您的~/sample_assembly_working_directory/report.txt文件或浏览~/sample_assembly_working_directory/Esch_coli_1_2015_000输出目录中的文件。~/sample_assembly_working_directory/Esch_coli_1_2015_000目录的内容也在~/sample_assembly_working_directory/Esch_coli_1_2015_000.tar.gz文件中压缩。将其移动到Windows机器,并按照https://github.com/i5K-KINBRE-script-share/Irys-scaffolding/blob/master/KSU_bioinfo_lab/assemble_XeonPhi/README.pdf文件中的说明查看IrysView中的对齐方式。以下步骤将很难完成,除非您已阅读README.md文件。
按照加载XMAP的说明,首先将原始计算机映射的XMAP文件导入到组装的基因组图中。这将在Esch_coli_1_2015_000/align_in_silico_xmap目录中。
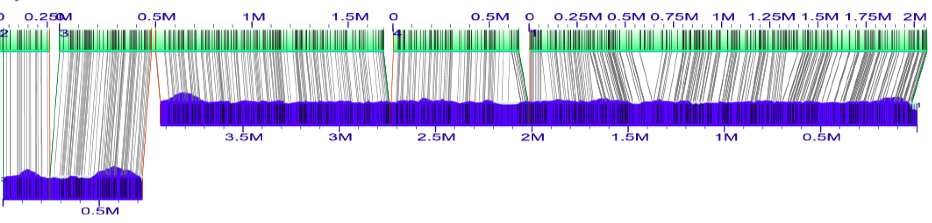
以上是第一个对齐方式的屏幕截图(在“silico map#2”,“silico map#3”,“silico map#4”,“silico map#1”中排序锚点“之后)。
接下来将计算机图中超级脚手架的XMAP文件导入到组装的基因组图中。这将在Esch_coli_1_2015_000/align_in_silico_super_scaffold_xmap目录中。
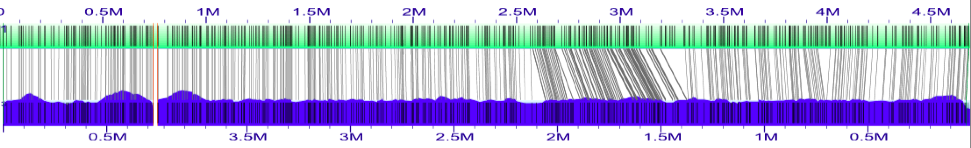
以上是第二次排列(在基准图上对齐的超级脚手架的超级脚手架)的屏幕截图。
接下来加载超薄脚架的重叠叠加料杯的BED文件在电脑地图中。这将是Esch_coli_1_2015_000/super_scaffold/Esch_coli_1_2015_000_20_40_15_90_2_superscaffold.fasta_contig.bed。还有一个BED文件的超级脚手架在电脑地图的空白,但这个样本基因组的差距非常小,因此在对齐中更难以查看Esch_coli_1_2015_000/super_scaffold/Esch_coli_1_2015_000_20_40_15_90_2_superscaffold.fasta_contig_gaps.bed。
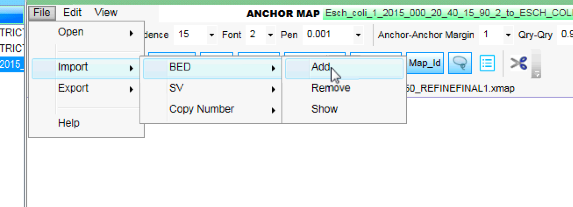 以上是您需要遵循的菜单的屏幕截图才能开始加载BED文件。
以上是您需要遵循的菜单的屏幕截图才能开始加载BED文件。
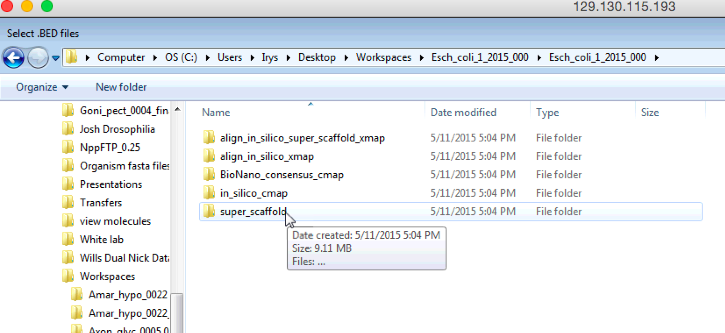 以上是您需要遵循的菜单的屏幕截图,以便找到超薄脚架在电子地图中的重叠盖BED文件。
以上是您需要遵循的菜单的屏幕截图,以便找到超薄脚架在电子地图中的重叠盖BED文件。
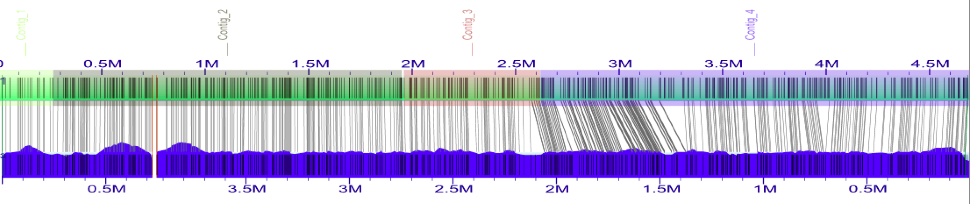 以上是加载了Contig BED文件的第二个对齐方式的屏幕截图。
以上是加载了Contig BED文件的第二个对齐方式的屏幕截图。
从查看最终的超脚手架对齐切换。sv_xmap将原始的电子地图从smaps目录中加载到组装的基因组图上。您可以通过在左侧的“比较图”窗口中突出显示其他对齐方式。按照README.md中的说明导入SMAP和合并的BED文件。
从屏幕底部的“Anchor”下拉列表中查看单个锚点时,您只会看到注释。锚点1是唯一具有预测结构变体的锚。查看注释通常需要根据README.md中的说明重新绘制图像。
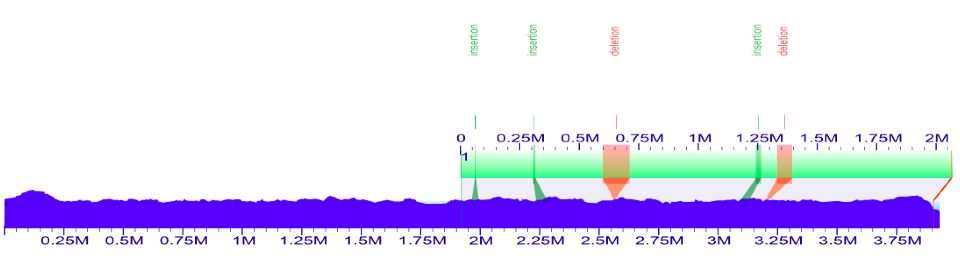 以上是在SV注释加载之后但在重绘以查看所有标签之前的计算机映射锚点#1的SV预测的屏幕截图。
以上是在SV注释加载之后但在重绘以查看所有标签之前的计算机映射锚点#1的SV预测的屏幕截图。
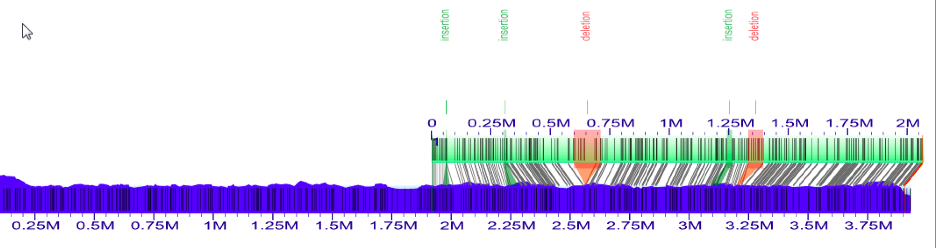 以上是SV注释加载后以及重新绘制以查看所有标签之后的计算机映射锚点#1的SV预测的屏幕截图。
以上是SV注释加载后以及重新绘制以查看所有标签之后的计算机映射锚点#1的SV预测的屏幕截图。