在之前的文章里,我主要讲了如下两个内容:(1) 认识Cell Hashing;(2): 使用Cell Ranger得到表达矩阵。相信大家已经知道了cell hashing与普通10X转录组的差异,以及使用cellranger得到表达矩阵。
这一篇讲如何使用Seurat的HTODemux函数,CiteFuse的crossSampleDoublets函数两种方法拆分表达矩阵(混了不同来源的细胞),最后还会略微比较一下两种方法得到的结果的差异。
HTODemux
这种方法的原理我在第一篇笔记中已经讲过,感兴趣的小伙伴可以看之前的文章。主要R代码如下:
library(Seurat)
library(ggplot2)
library(tidyverse)
args <- commandArgs(TRUE)
加载R包,导入外部参数,args[1]表示样本名称,args[2]表示ensembl_ID和基因symbol对应关系的文本文件,前面得到的表达矩阵行名是ensembl_ID,为了在后续可视化的时候更省事,建议在这一步更换基因名称。
df <- read.table(paste(args[1],".mat.count.txt",sep = ""),header = T,row.names = 1) #df的行数包括基因和tag
colnames(df) <- str_replace(colnames(df),"\.1","")
ensembl_symbol <- read.table(args[2],header = F,row.names = 1,stringsAsFactors = F)
df1 <- df[intersect(rownames(ensembl_symbol),rownames(df)),] #提取出基因表达矩阵
df2 <- df[setdiff(rownames(df),rownames(ensembl_symbol)),] #提取出tag表达矩阵
rownames(df1) <- ensembl_symbol[rownames(df1),] #更换基因表达矩阵的行名
接下来利用df2数据框的信息拆分,df2行为tag,列为cellular barcode
cellhash <- CreateSeuratObject(counts = df2,project = "cell_hashing", assay = "HTO")
cellhash <- NormalizeData(cellhash, assay = "HTO", normalization.method = "CLR")
cellhash <- HTODemux(cellhash, assay = "HTO", positive.quantile = 0.85)
最后一步就是拆分,第一篇笔记说过,positive.quantile参数表示在拟合负二项分布之后使用什么分位数来判断UMI是相对大还是相对小,默认值是0.99,实际处理时,发现这个值可能并不合理,比如最终拆分出来的有效细胞数、不同来源细胞数比例和预期差别很大,再比如从图形上看,明显不对(下文有图形说明)。
这一步之后,每一个CB都会带上一个标签,比如我的数据只有两个样本来源,标签会有这4种:Negative、tag6_tag7、tag6、tag7,前面两个表示空液滴、(跨样本的)doublet。
Idents(cellhash) <- "HTO_classification"
FeatureScatter(cellhash, feature1 = paste("hto_",rownames(cellhash)[1],sep=""),
feature2 = paste("hto_",rownames(cellhash)[2],sep = ""),slot = "counts")
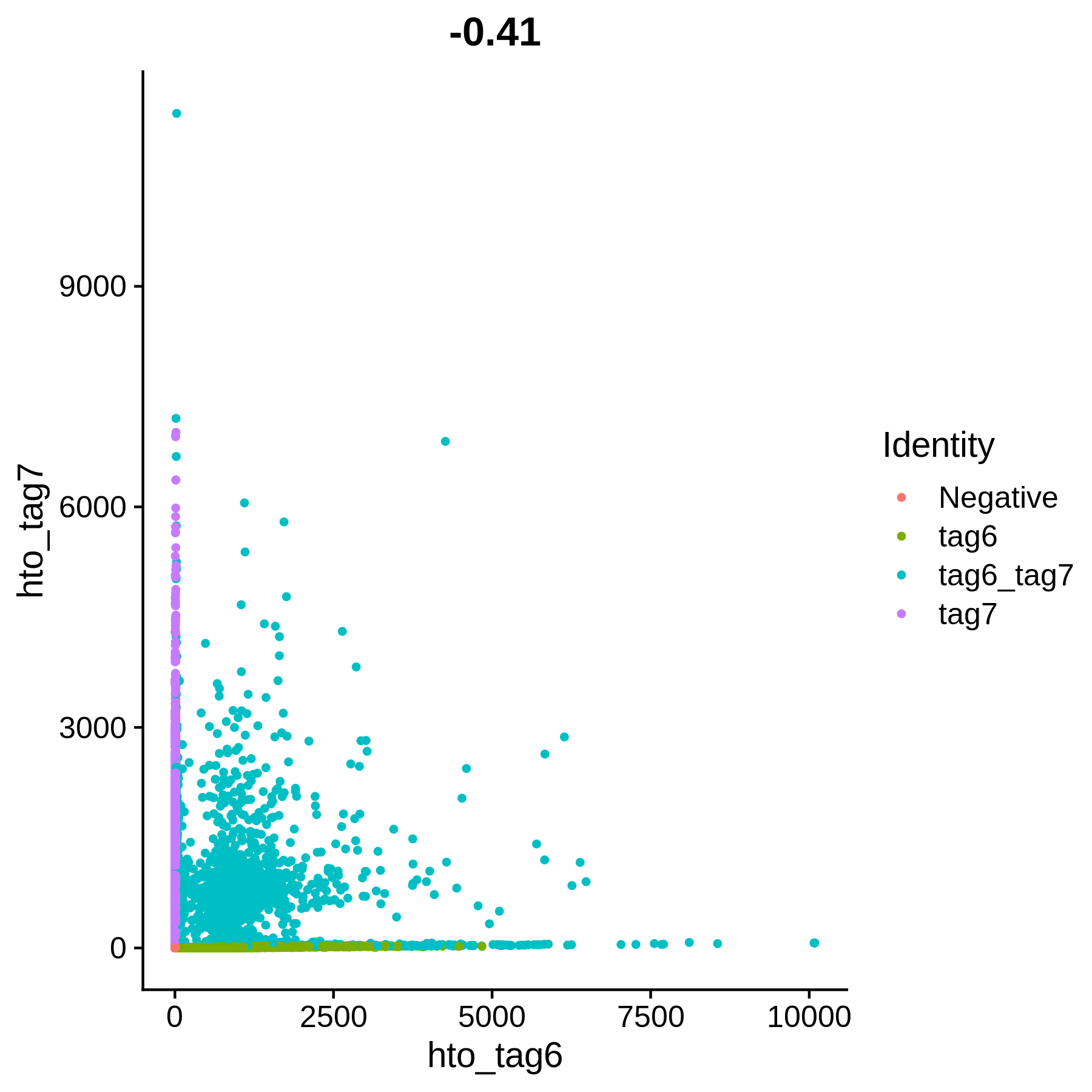
HTOHeatmap(cellhash, assay = "HTO")

上面两个图,可以用来检验拆分的质量,第一张每个点的横纵坐标表示每个CB两个tag的UMI,第二张图的每一列表示每个CB两个tag的标准化之后的表达量。
然后根据每个CB的标签提取出有效的singlet就可以了。
small_df1 <- df1[,colnames(cellhash)[cellhash$HTO_classification==rownames(cellhash)[1]]]
write.table(small_df1,paste(args[1],"_",rownames(cellhash)[1],".mat.count.txt",sep = ""),quote = F,row.names = T,col.names = T,sep=" ")
small_df2 <- df1[,colnames(cellhash)[cellhash$HTO_classification==rownames(cellhash)[2]]]
write.table(small_df2,paste(args[1],"_",rownames(cellhash)[2],".mat.count.txt",sep = ""),quote = F,row.names = T,col.names = T,sep=" ")
除了上面两种Seurat自带图形,下面两种图形也很有参考意义,代码就先不放了,如有需要可以在公众号后台小窗我。
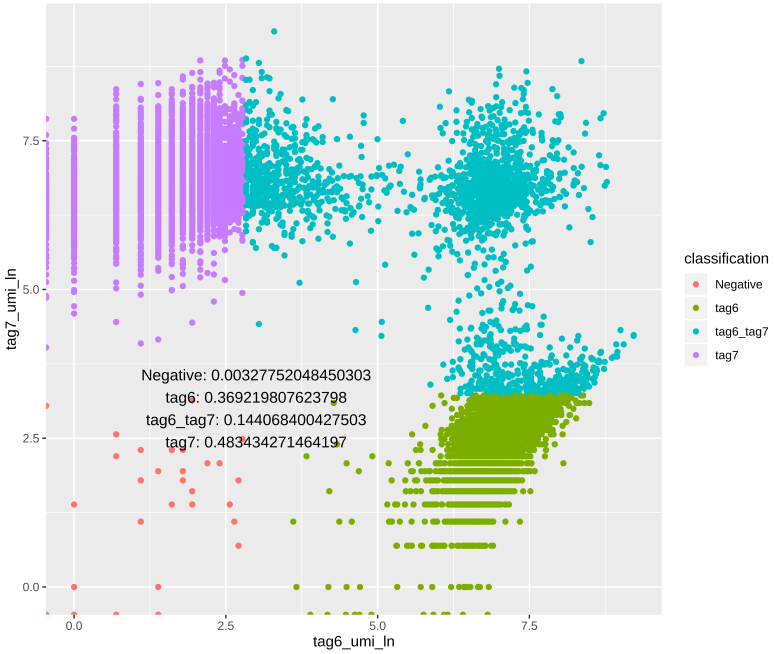

将UMI取对数之后做图,可以从另一个角度看结果,可以看到右上角被HTODemux认定为doublet的CB,像是包含了本应该是singlet的CB。我尝试过positive.quantile用默认值0.99,这种现象会更明显,所以我觉得在做这一步的时候,可以画画这个图,选择一个适中的positive.quantile值。
crossSampleDoublets
CiteFuse包在做这一步的时候,是从取对数之后的UMI矩阵开始的,分别从两个维度拟合正态分布,因此最终得到的结果在散点图上,比上一种方法更说得过去。示意图如下:

具体使用的R代码如下:
library(tidyverse)
library(ggplot2)
library(SingleCellExperiment)
library(CiteFuse)
args <- commandArgs(TRUE)
allexp <- read.table(paste(args[1],".mat.count.txt",sep = ""),header = T,row.names = 1)
colnames(allexp) <- str_replace(colnames(allexp),"\.1","")
allexp_sce <- preprocessing(exprsMat = as.matrix(allexp)) #生成特定的对象
is.HTO <- grepl("^tag[123678]", rownames(allexp_sce)) #根据自己的tag命名修改正则表达式
allexp_sce <- splitAltExps(allexp_sce, ifelse(is.HTO, "HTO", "gene")) #给每一行加一个标签,HTO或者gene
allexp_sce=normaliseExprs(allexp_sce, altExp_name = "HTO", exprs_value = "counts",transform = c("log")) #仅针对HTO行,取对数
allexp_sce=crossSampleDoublets(allexp_sce,altExp_name = "HTO",totalExp_threshold = 10)
最后一行就是拆分关键步骤,会给每个CB一个标签,totalExp_threshold表示只会保留表达数大于10的CB。
ensembl_symbol <- read.table("/ref/10x/Ensembl_symbol_new.txt",header = F,row.names = 1,stri
ngsAsFactors = F)
df1 <- allexp[intersect(rownames(ensembl_symbol),rownames(allexp)),]
df2 <- allexp[setdiff(rownames(allexp),rownames(ensembl_symbol)),]
rownames(df1) <- ensembl_symbol[rownames(df1),]
tmp1=as.data.frame(t(df2))
tmp2=as.data.frame(allexp_sce$doubletClassify_between_label)
colnames(tmp2)="anno"
tmp2$anno=as.character(tmp2$anno)
crossSampleDoublets返回的标签不容易识别,比如1、2,还需要重新更换名称,如下
for (i in seq(1,length(rownames(tmp2)),1)) {
for (j in seq(1,length(colnames(tmp2)),1)) {
if (tmp2[i,j] == "1") {
tmp2[i,j] = colnames(tmp1)[1]
}
if (tmp2[i,j] == "2") {
tmp2[i,j] = colnames(tmp1)[2]
}
if (tmp2[i,j] == "doublet/multiplet") {
tmp2[i,j] = "doublet"
}
}
}
df_point=cbind(tmp1,tmp2)
colnames(df_point)=c("taga","tagb","anno")
这一步之后就能根据tag标签画散点图,以及提取想要的矩阵了。
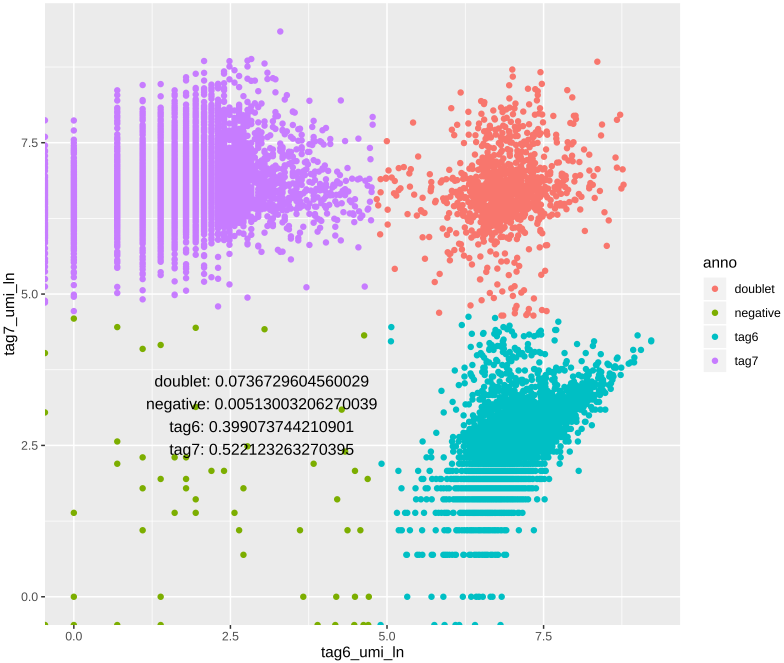
实际处理中,上面两种方法我都用了,最后选了二者交集的CB来提取矩阵(相对保险的做法)。这一步在cell hashing数据的处理中可以说是相当重要了,如果拆分质量不过关,错误地将不同来源的细胞划分到一个矩阵中,对后续分析结果影响很大。
上述代码只呈现了拆分的关键步骤,详细的画图代码没有放上来,如果需要可以在微信后台私信我。
因水平有限,有错误的地方,欢迎批评指正!
