上一篇已经讲解了Seurat标准流程,推文的最后,注意到了不同样本之间的表达数据是存在批次效应的,就像下图这样,有些是可以聚到一起的亚群,却出现了不同样本分开/偏移的情况,比如第3群,这种就是批次效应:

接下来我会介绍Seurat v3的标准整合流程、Seurat结合Harmony 的整合流程,仍然使用上一个数据集
1. Seurat v3的标准整合流程
对于不同样本先分别运行Seurat的标准流程到找Variable基因这一步
library(Seurat)
library(tidyverse)
### testA ----
testA.seu=CreateSeuratObject(counts = testA)
testA.seu <- NormalizeData(testA.seu, normalization.method = "LogNormalize", scale.factor = 10000)
testA.seu <- FindVariableFeatures(testA.seu, selection.method = "vst", nfeatures = 2000)
### testB ----
testB.seu=CreateSeuratObject(counts = testB)
testB.seu <- NormalizeData(testB.seu, normalization.method = "LogNormalize", scale.factor = 10000)
testB.seu <- FindVariableFeatures(testB.seu, selection.method = "vst", nfeatures = 2000)
然后就是主要的整合步骤,object.list参数是由多个Seurat对象构成的列表,如下:
### Integration ----
testAB.anchors <- FindIntegrationAnchors(object.list = list(testA.seu,testB.seu), dims = 1:20)
testAB.integrated <- IntegrateData(anchorset = testAB.anchors, dims = 1:20)
需要注意的是:上面的整合步骤相对于harmony整合方法,对于较大的数据集(几万个细胞),非常消耗内存和时间;当存在某一个Seurat对象细胞数很少(印象中200以下这样子),会报错,这时建议用第二种整合方法
这一步之后就多了一个整合后的assay(原先有一个RNA的assay),整合前后的数据分别存储在这两个assay中
> testAB.integrated
An object of class Seurat
35538 features across 6746 samples within 2 assays
Active assay: integrated (2000 features)
1 other assay present: RNA
> dim(testAB.integrated[["RNA"]]@counts)
[1] 33538 6746
> dim(testAB.integrated[["RNA"]]@data)
[1] 33538 6746
> dim(testAB.integrated[["integrated"]]@counts) #因为是从RNA这个assay的data矩阵开始整合的,所以这个矩阵为空
[1] 0 0
> dim(testAB.integrated[["integrated"]]@data)
[1] 2000 6746
后续仍然是标准流程,基于上面得到的整合data矩阵
DefaultAssay(testAB.integrated) <- "integrated"
# Run the standard workflow for visualization and clustering
testAB.integrated <- ScaleData(testAB.integrated, features = rownames(testAB.integrated))
testAB.integrated <- RunPCA(testAB.integrated, npcs = 50, verbose = FALSE)
testAB.integrated <- FindNeighbors(testAB.integrated, dims = 1:30)
testAB.integrated <- FindClusters(testAB.integrated, resolution = 0.5)
testAB.integrated <- RunUMAP(testAB.integrated, dims = 1:30)
testAB.integrated <- RunTSNE(testAB.integrated, dims = 1:30)
看一下去除批次效应之后的结果
library(cowplot)
testAB.integrated$patient=str_replace(testAB.integrated$orig.ident,"_.*$","")
p1 <- DimPlot(testAB.integrated, reduction = "tsne", group.by = "patient", pt.size=0.5)+theme(
axis.line = element_blank(),
axis.ticks = element_blank(),axis.text = element_blank()
)
p2 <- DimPlot(testAB.integrated, reduction = "tsne", group.by = "ident", pt.size=0.5, label = TRUE,repel = TRUE)+theme(
axis.line = element_blank(),
axis.ticks = element_blank(),axis.text = element_blank()
)
fig_tsne <- plot_grid(p1, p2, labels = c('patient','ident'),align = "v",ncol = 2)
ggsave(filename = "tsne2.pdf", plot = fig_tsne, device = 'pdf', width = 27, height = 12, units = 'cm')

可以看到,不同样本的细胞基本都分散均匀了,结果还不错
2. Seurat结合Harmony的整合流程
这种整合方法很简单,而且占内存少,速度快
library(harmony)
testdf=cbind(testA,testB)
test.seu <- CreateSeuratObject(counts = testdf) %>%
Seurat::NormalizeData() %>%
FindVariableFeatures(selection.method = "vst", nfeatures = 2000) %>%
ScaleData()
test.seu <- RunPCA(test.seu, npcs = 50, verbose = FALSE)
test.seu@meta.data$patient=str_replace(test.seu$orig.ident,"_.*$","")
先运行Seurat标准流程到PCA这一步,然后就是Harmony整合,可以简单把这一步理解为一种新的降维
test.seu=test.seu %>% RunHarmony("patient", plot_convergence = TRUE)
> test.seu
An object of class Seurat
33538 features across 6746 samples within 1 assay
Active assay: RNA (33538 features)
2 dimensional reductions calculated: pca, harmony
接着就是常规聚类降维,都是基于Harmony的Embeddings矩阵
test.seu <- test.seu %>%
RunUMAP(reduction = "harmony", dims = 1:30) %>%
FindNeighbors(reduction = "harmony", dims = 1:30) %>%
FindClusters(resolution = 0.5) %>%
identity()
test.seu <- test.seu %>%
RunTSNE(reduction = "harmony", dims = 1:30)
看看效果
p3 <- DimPlot(test.seu, reduction = "tsne", group.by = "patient", pt.size=0.5)+theme(
axis.line = element_blank(),
axis.ticks = element_blank(),axis.text = element_blank()
)
p4 <- DimPlot(test.seu, reduction = "tsne", group.by = "ident", pt.size=0.5, label = TRUE,repel = TRUE)+theme(
axis.line = element_blank(),
axis.ticks = element_blank(),axis.text = element_blank()
)
fig_tsne <- plot_grid(p3, p4, labels = c('patient','ident'),align = "v",ncol = 2)
ggsave(filename = "tsne3.pdf", plot = fig_tsne, device = 'pdf', width = 27, height = 12, units = 'cm')
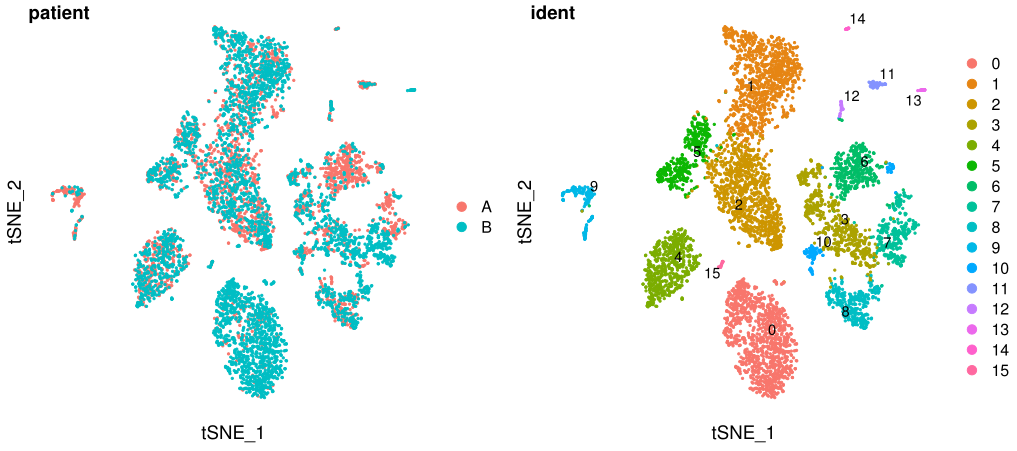
看起来也挺好的~
对于异质性很大的不同数据集,比如不同病人的肿瘤细胞,考虑到生物学差异已经远远大于批次效应,这时不应该再进行批次效应的去除,不然内在的生物学差异也会抹掉。尽管也在文献里面看到过对肿瘤细胞去批次的,但我始终觉得这样做是错的。不知道大家是怎么看的?
最后还有一个有用的小技巧,在降维之后,我们可以用DimPlot()函数把结果画在二维平面上,那这些坐标信息存储在哪儿呢?存储在Embeddings(test.seu,"pca")矩阵中,harmony/tsne/umap同理。有时候我们可以把坐标和meta@data的信息提取出来,用ggplot2画图,方便修改。
> Embeddings(test.seu,"tsne")[1:4,1:2]
tSNE_1 tSNE_2
A_AAACCCAAGGGTCACA -6.774032 -25.08772
A_AAACCCAAGTATAACG -5.294804 39.77550
A_AAACCCAGTCTCTCAC 30.600943 31.28015
A_AAACCCAGTGAGTCAG -32.432794 -14.62455
因水平有限,有错误的地方,欢迎批评指正!