前面已经讲解了:
单细胞分析实录(1): 认识Cell Hashing
单细胞分析实录(2): 使用Cell Ranger得到表达矩阵
单细胞分析实录(3): Cell Hashing数据拆分
单细胞分析实录(4): doublet检测
单细胞分析实录(5): Seurat标准流程
单细胞分析实录(6): 去除批次效应/整合数据
这一节我们进入细胞类型注释的学习,总的来说,两条路线,手动注释和软件注释。我在实际处理的过程中,主要还是手动注释,软件的注释结果只作为一个参考,来辅助证实手动注释的结果是准确无误的。
相信看过几篇单细胞文献的小伙伴基本都会知道几种常见细胞大类的marker,我们在注释的时候用这些marker就可以基本确定细胞大类了。另外,以SingleR为例,对于细胞大类的注释还算准确,当然也没有到很准确的程度,我试过更换SingleR里面不同的参考集,最后得到的大类注释结果一致性不到80%。对于细胞小类的注释,软件就更加无法胜任了,我几乎没有看过高分的文献会用SingleR的小类注释结果。当然不排除随着单细胞研究的普及和深入,以后能有更准确的软件出现。
接下来,我以上一节的数据为例,走一遍我的分析流程。
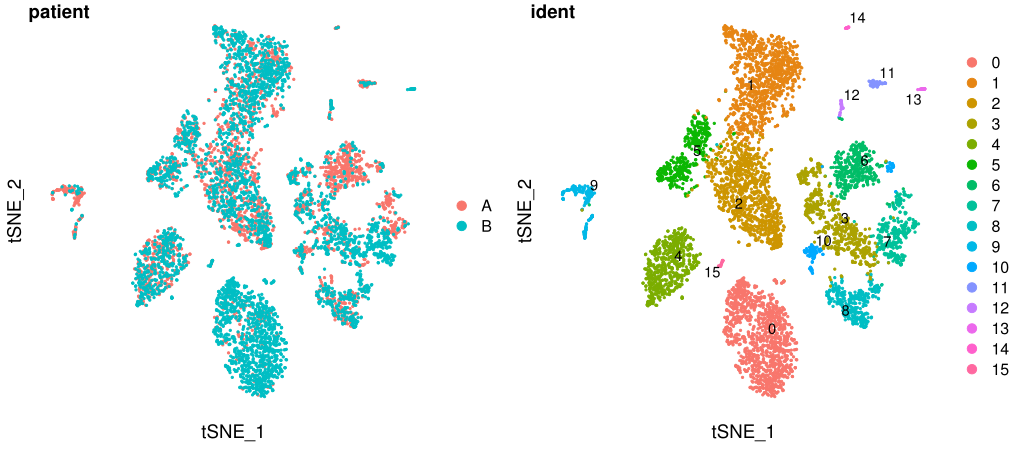
之前的数据呈现出了16个cluster,至于resolution参数的选择,我的原则是能在降维图上分开的细胞亚群,能有它自己的cluster label,并且成团较好,较紧致的一群细胞没有必要再强行分群(比如上图的第4群)。
这16个cluster不一定都会用到,因为doublet、细胞数太少等原因,可能还得去掉。
我推荐用常见的marker先区分一下,大概能知道cluster对应什么类型。这里用到的marker都是在文献里面经常出现的,我自己也整理了一份更全一点的细胞类型marker清单,有需要的可以在公众号后台说明。
celltype_marker=c(
"EPCAM",#上皮细胞 epithelial
"PECAM1",#内皮细胞 endothelial
"COL3A1",#成纤维细胞 fibroblasts
"CD163","AIF1",#髓系细胞 myeloid
"CD79A",#B细胞
"JCHAIN",#浆细胞 plasma cell
"CD3D","CD8A","CD4",#T细胞
"GNLY","NKG7",#NK细胞
"PTPRC"#免疫细胞
)
VlnPlot(test.seu,features = celltype_marker,pt.size = 0,ncol = 2)
ggsave(filename = "marker.png",device = "png",width = 44,height = 33,units = "cm")
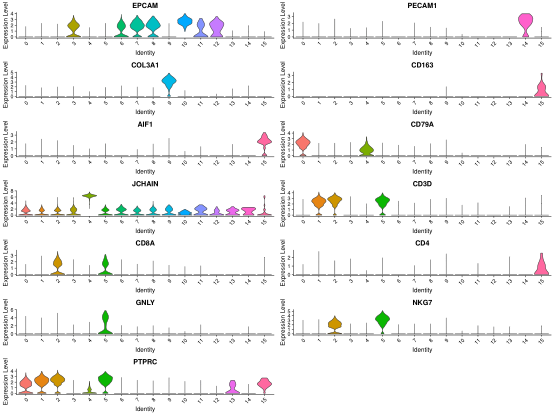
结合对应的关系,初步确认细胞类型如下,需要说明的是,NK细胞和T细胞不是很能区分开,一些文献也是直接把这两个当做一个大群来做的,此处我根据GNLY基因确定第5个cluster为NK细胞,是否正确我们后面再看。
#免疫细胞
0: B_cell
4: Plasma_cell
1 2: T_cell
5: NK_cell
13: Unknown
#非免疫细胞
3 6 7 8 10 11 12: Epithelial
14: Endothelial
9: Fibroblasts
#doublet
15: Doublet (Myeloid+CD4)
以上根据经典的marker初步确定了细胞大类,接着我们找差异基因,看看找出来的每一个cluster的差异基因是不是和前面鉴定的类型一致。
1. 找差异基因
markers <- FindAllMarkers(test.seu, logfc.threshold = 0.25, min.pct = 0.1,
only.pos = TRUE, test.use = "wilcox")
markers_df = markers %>% group_by(cluster) %>% top_n(n = 500, wt = avg_logFC)
write.table(markers_df,file="test.seu_0.5_logfc0.25_markers500.txt",quote=F,sep=" ",row.names=F,col.names=T)
FindAllMarkers()这个函数会将cluster中的某一类和剩下的那些类比较,来找差异基因。与其对应的有个FindMarkers()函数,可以指定哪两个cluster对比。logfc.threshold表示logfc的阈值,这里有两个地方需要注意:一是Seurat里面的logfc计算公式很特别,并不是我们平常在bulk里面那样算均值,相除,求log,但其实也不要纠结怎么算的,只需知道这是表示倍数的一个指标即可;二是如果想画火山图,这个阈值可以设为0,不然最后画出来的火山图中间会缺一段,我们完全可以把全部基因先拿出来,画火山图,再根据p value, logFC这些阈值自己过滤。
min.pct表示基因在多少细胞中表达的阈值,only.pos = TRUE表示只求高表达的基因,test.use表示检测差异基因所用的方法。
第2行代码,根据avg_logFC这一列把前500个差异基因取出来,小于500个时,有多少保留多少。
事实上,不必在意我们保留多少个差异基因,实际用到的,也就前面十几(几十)个基因。
最终得到的差异基因list如下:
> head(markers_df)
# A tibble: 6 x 7
# Groups: cluster [1]
p_val avg_logFC pct.1 pct.2 p_val_adj cluster gene
<dbl> <dbl> <dbl> <dbl> <dbl> <fct> <chr>
1 0 3.58 0.999 0.114 0 0 HLA-DRA
2 0 2.72 1 0.331 0 0 CD74
3 0 2.59 0.983 0.169 0 0 HLA-DPA1
4 0 2.50 0.985 0.148 0 0 HLA-DPB1
然后,我们可以根据每一个cluster排在前面的基因验证前面鉴定的结果是否正确。我把刚刚得到的的表格看一下,基本符合前面的鉴定。
2. SingleR注释
我刚开始用SingleR是在19年的年底,现在使用方法可能有一些变化了。因为Single在注释细胞的过程中,会用到一些参考数据集,我们可以把这些数据集保存下来,下一次使用就可以直接加载,省去了重新下载参考集的时间。
library(celldex)
hpca.se <- HumanPrimaryCellAtlasData()
hpca.se
## class: SummarizedExperiment
## dim: 19363 713
## metadata(0):
## assays(1): logcounts
## rownames(19363): A1BG A1BG-AS1 ... ZZEF1 ZZZ3
## rowData names(0):
## colnames(713): GSM112490 GSM112491 ... GSM92233 GSM92234
## colData names(3): label.main label.fine label.ont
save(hpca.se,file="/ref/singler/HPCA/hpca.se.RData")
注意参考集里面是logcounts矩阵,后面对于单细胞数据集也要做类似的处理。
导入UMI count矩阵
library(SingleR)
library(scater)
library(SummarizedExperiment)
test.count=as.data.frame(test.seu[["RNA"]]@counts)
导入参考集,保证两个数据集的基因相同,然后log处理
load(file="hpca.se.RData")
common_hpca <- intersect(rownames(test.count), rownames(hpca.se))
hpca.se <- hpca.se[common_hpca,]
test.count_forhpca <- test.count[common_hpca,]
test.count_forhpca.se <- SummarizedExperiment(assays=list(counts=test.count_forhpca))
test.count_forhpca.se <- logNormCounts(test.count_forhpca.se)
接下来是注释步骤,在这一步里,我只用了main大类注释,还有一个fine小类注释,我就不演示了,因为我觉得小类注释不太准。
###main
pred.main.hpca <- SingleR(test = test.count_forhpca.se, ref = hpca.se, labels = hpca.se$label.main)
result_main_hpca <- as.data.frame(pred.main.hpca$labels)
result_main_hpca$CB <- rownames(pred.main.hpca)
colnames(result_main_hpca) <- c('HPCA_Main', 'CB')
write.table(result_main_hpca, file = "HPCA_Main.txt", sep = ' ', row.names = FALSE, col.names = TRUE, quote = FALSE) #保存下来,方便以后调用
得到的结果是这样的,每个CB都有一个label
> head(result_main_hpca)
HPCA_Main CB
1 B_cell A_AAACCCAAGGGTCACA
2 T_cells A_AAACCCAAGTATAACG
3 Epithelial_cells A_AAACCCAGTCTCTCAC
4 B_cell A_AAACCCAGTGAGTCAG
5 T_cells A_AAACCCAGTGGCACTC
6 T_cells A_AAACGAAAGCCAGAGT
我们接下来要把这个结果整合到test.seu@meta.data中,然后画tsne/umap展示一下
test.seu@meta.data$CB=rownames(test.seu@meta.data)
test.seu@meta.data=merge(test.seu@meta.data,result_main_hpca,by="CB")
rownames(test.seu@meta.data)=test.seu@meta.data$CB
p5 <- DimPlot(test.seu, reduction = "tsne", group.by = "HPCA_Main", pt.size=0.5)+theme(
axis.line = element_blank(),
axis.ticks = element_blank(),axis.text = element_blank()
)
p6 <- DimPlot(test.seu, reduction = "tsne", group.by = "ident", pt.size=0.5, label = TRUE,repel = TRUE)+theme(
axis.line = element_blank(),
axis.ticks = element_blank(),axis.text = element_blank()
)
fig_tsne <- plot_grid(p6, p5, labels = c('ident','HPCA_Main'),rel_widths = c(2,3))
ggsave(filename = "tsne4.pdf", plot = fig_tsne, device = 'pdf', width = 36, height = 12, units = 'cm')
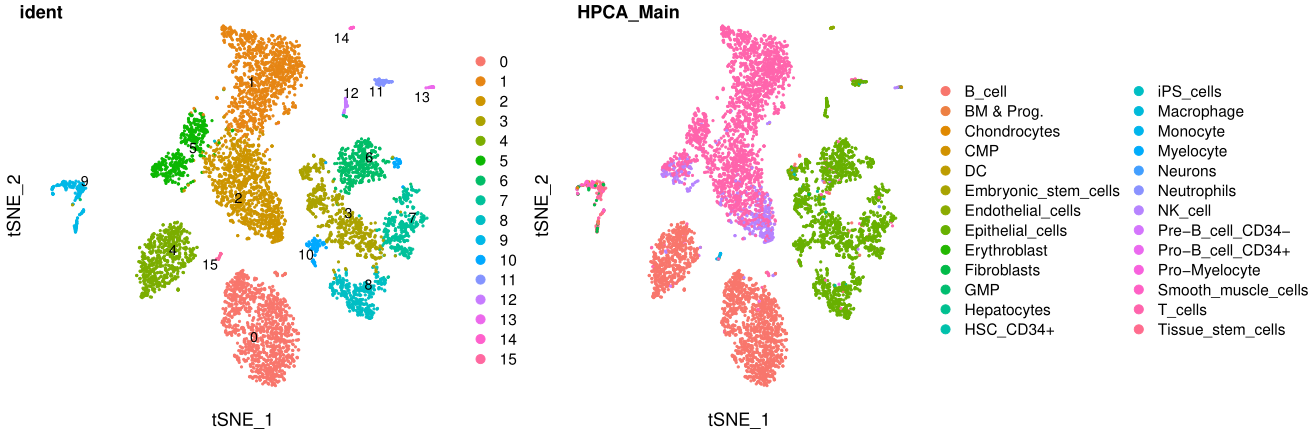
整体看上去也是符合最初的鉴定的
到这儿,算是把大类鉴定了,最后我们把注释信息添加上去
B_cell=c(0)
Plasma_cell=c(4)
T_cell=c(1,2)
NK_cell=c(5)
Unknown=c(13)
Epithelial=c(3, 6, 7, 8, 10, 11, 12)
Endothelial=c(14)
Fibroblasts=c(9)
Doublet=c(15)
current.cluster.ids <- c(B_cell,
Plasma_cell,
T_cell,
NK_cell,
Unknown,
Epithelial,
Endothelial,
Fibroblasts,
Doublet)
new.cluster.ids <- c(rep("B_cell",length(B_cell)),
rep("Plasma_cell",length(Plasma_cell)),
rep("T_cell",length(T_cell)),
rep("NK_cell",length(NK_cell)),
rep("Unknown",length(Unknown)),
rep("Epithelial",length(Epithelial)),
rep("Endothelial",length(Endothelial)),
rep("Fibroblasts",length(Fibroblasts)),
rep("Doublet",length(Doublet))
)
test.seu@meta.data$celltype <- plyr::mapvalues(x = as.integer(as.character(test.seu@meta.data$seurat_clusters)), from = current.cluster.ids, to = new.cluster.ids)
新的test.seu@meta.data是这样的:
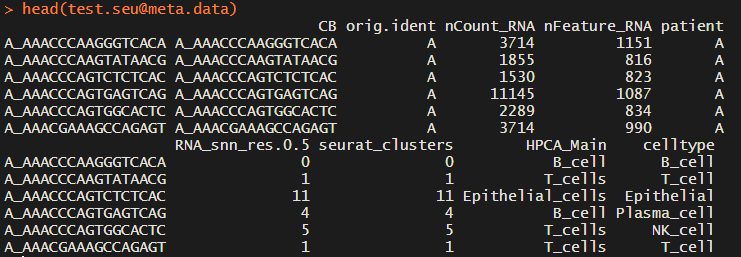
统计一下各种细胞的数目
> table(test.seu@meta.data$celltype)
B_cell Doublet Endothelial Epithelial Fibroblasts NK_cell Plasma_cell
1188 22 27 2023 205 441 638
T_cell Unknown
2167 35
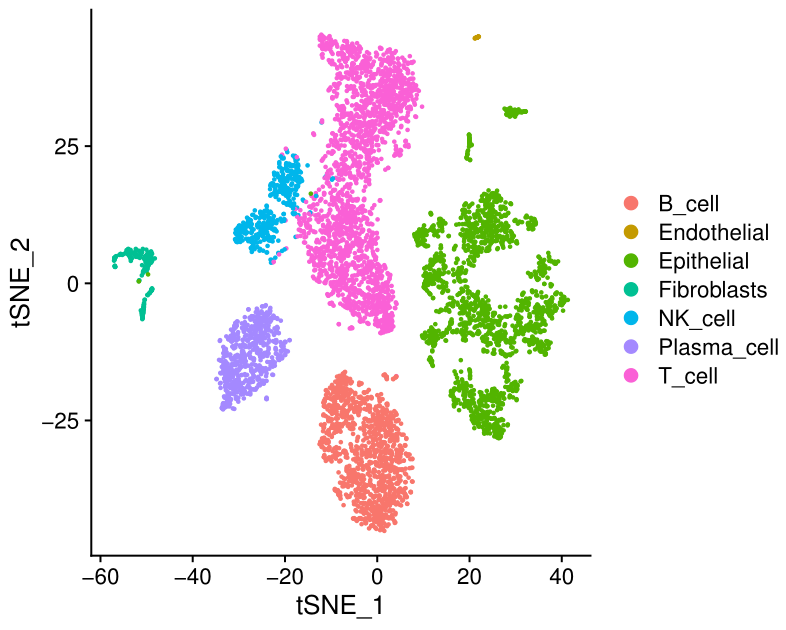
我们把Doublet和Unknown去掉,画最后的tsne图
plotCB=as.data.frame(test.seu@meta.data%>%filter(seurat_clusters!="13" &
seurat_clusters!="15"))[,"CB"]
DimPlot(test.seu, reduction = "tsne", group.by = "celltype", pt.size=0.5,cells = plotCB)
ggsave(filename = "tsne5.pdf", width = 15, height = 12, units = 'cm')
saveRDS(test.seu,file = "test.seu.rds") #保存test.seu对象,下次可以直接调用,不用重复以上步骤
最后分享一个找差异基因的小技巧,有时候,我们希望对自己定义的类群找差异基因,可以用下面这种方法
Idents(test.seu)="celltype"
markers2 <- FindAllMarkers(test.seu, logfc.threshold = 0.25, min.pct = 0.1,
only.pos = TRUE)

因水平有限,有错误的地方,欢迎批评指正!