这些三代全长转录组测序的相关问题,可以帮到爱学习的我哦
1.什么是三代全长转录组测序
三代全长转录组测序,即利用PacBio三代测序平台对某一物种的mRNA进行测序研究。它以平均超长读长10-15kb的优势、结合多片段文库筛选技术,实现了无需拼接的转录本分析,克服了传统二代转录组Unigene拼接较短、转录本结构不完整的缺陷,也由于其可直接获得单个RNA分子从5’端到3’端的高质量全部转录组信息而得名。
2.为什么要做全长转录组测序?
转录本非常多样和复杂,绝大多数基因不符合“一基因一转录本”的模式,这些基因往往存在多种剪切形式。通过二代测序,我们可以很准确地进行基因的表达及定量的研究,但是受限于读长的限制,不能得到全长转录本的信息。
基于二代测序平台的转录组产品,首先是把RNA打成小的短片断进行测序,然后再通过生物信息的方法进行拼接,将拼接后的序列交付给客户。但是基于二代测序平台的转录组,由于读长的限制(PE150),在转录本组装的过程中会存在较多的嵌合体,并且不能准确地得到完整转录本的信息,从而会大大降低表达量、可变剪接、基因融合等分析的准确性。图1. 二代和三代转录组测序原理及读长对比
目前基于PacBio的单分子实时测序技术,目前平均读长已经达到10Kb以上,最长可达80Kb,转录组测序不再需要组装,就可以直接得到全长转录本的信息。
3.二代与三代转录组相比两者分别有哪些优劣势?三代转录组具体优势可否说明?
表1. 二代和三代转录组测序优劣势对比
从上述对比表格可看出,两种转录组测序技术互有优劣势,所以在给各位老师在设计课题时,建议老师二+三代转录组测序技术同时使用,保证结构准确性、序列完整性及序列表达量准确性,达到数据的最优利用效果以及性价比最高。
三代转录组具体优势说明如下:
a.超长读长(平均读长10-15K,最长读长80K),可一次将真核生物的全长转录本信息读取完整;
b.无需进行片段打断和拼接,避免出现组装错误;
c.基于全长转录组测序得到的完整准确的转录本信息,结合二代数据,方便识别特异性表达且做更加精确的基因和转录本表达定量。
d.针对有参考基因组的物种,全长转录组信息可以纠正基因组的错误组装、更准确地发现新的转录本和基因、分析基因融合事件等。
e.无需链特异性建库,全长转录组测序可直接获取正义链、反义链及部分LncRNA信息。
图2. 三代全长转录组测序优势概览
4.哪些物种适合做三代全长转录组测序?获得这些全长mRNA信息有何用处?
无参考基因组物种和有参考基因组物种均适用。
a.对于没有参考基因组的物种
由于基因组测序成本高,缺乏基因组参考信息在很大程度上限制了对物种的深入研究。通过三代全长转录组测序来构建物种Unigene库,无需进行序列组装,就可以获得该物种转录组水平的参考序列(转录组水平的参考基因组),为后续研究提供很好的遗传信息基础。
获得这些全长转录本信息,可以更准确地进行CDS和SSR分析。如果有同一批样本的二代数据,不但可以提高三代测序数据的利用率,同时可以对这些全长的转录本进行更精准的定量分析。
b.对于有不完善参考基因组的物种
参考基因组组装不完善是普遍存在的问题,特别是多倍体这类物种,给科研工作带来了极大阻碍。参考基因组组装不完善,用二代测序会导致reads比对率低,基因表达定量不准确的问题。用全长转录组测序技术可直接获得转录本全长序列,再结合二代测序,会使定量更准确,数据利用率更高,同时基于全长转录组数据,可以优化基因结构,进而辅助基因组组装和注释。
c.对于具有较好参考基因组的物种
利用三代全长转录组测序获得的信息是生物体内直接存在的,比基于参考基因组预测到的转录组信息更准确,同时也可准确鉴定基因的可变剪接、融合基因、基因家族和非编码RNA等信息。
如果有同一样本的二代数据,不但可以提高三代数据的利用率,同时还可以深入研究某基因可变剪接形成的不同转录本的表达差异。可以确定不同发育阶段或不同处理情况下,该基因中高表达转录本以及低表达转录本。不同样品的融合基因和等位基因差异,也同样可以分析。
需要注意的是全长转录组测序只能得到转录本全长序列,不可进行基因表达定量。
5. 全长转录组测序那么贵,如何更大程度上降低测序成本?
由于转录组信息呈动态变化且存在组织差异,单一组织得到的全长转录本对该物种其他部位组织可能不是很全面或不太适用,所以用一个物种不同部位组织混样进行高深度测序(针对不同要求及目的,推荐8G、10G和12G等),会得到比较理想的参考转录本库信息,也是降低测序成本的理想方法。
6. 三代全长转录组测序如何选择测序样本?
总体原则是根据研究目的进行选择,举例说明如下:
a.单个三代转录组项目:
① 如果想要获得该物种相对全面的转录本信息,建议对该物种的不同部位混合取样;
② 如果只想研究某个特定的组织部位,建议在不同发育时期对特定组织部位进行取样;
b. 二+三代转录组混合策略项目:
三代转录组与二代转录组测序取样部位或时期相对应的同一批样品,等量RNA混样测序;
c. 多个三代转录组样品项目:
如果想要研究某物种胁迫处理(其他生物或非生物胁迫都适用)前后变化,建议取对照和处理组(至少两个样品)进行对比分析;
① 全长转录组混样测序为了保证数据来源的均一性,一定要等量RNA混合测序,而非等量样品混合抽取RNA再测序。
② 随着三代转录组测序成本逐步下降,多个三代转录组样品测序的常规时代也即将到来。
7. 全长转录本数据量和文库类型如何确定?
推荐数据量大小需依据物种的复杂程度、基因大小及研究目的来确定。根据已有的项目经验、数据库信息及文章中报道,我们详细推荐如下:
表2. 推荐性全长转录本测序数据量和文库类型
注:对于全长转录组测序,数据量并不是固定的,针对同一物种同一研究目的,测序数据量越多,检测到的全长转录本也会越全面。
8. 全长转录组测序为什么要建3-4个分段文库?不同文库数据产出比例如何?
构建分段文库,是由PacBio平台测序原理所决定。在三代转录组测序过程中,构建好的全长文库需要loading到测序小孔——零模波导孔(ZMWs)中,由于mRNA长度不同,在loading的过程中会出现一定的loading bias,即测序小孔会优先被长度较短的片段占据,每个测序小孔只能容纳一个文库分子,而大部分长片段则没有测到。因此为尽量降低loading bias的影响,需要根据测序物种mRNA的长度进行分段,使一个文库中的序列长度控制在一个较窄的范围内。故构建分级文库越多,也会得到更全面的全长转录本。
全长转录组测序一般推荐至少构建三种文库类型,1-2Kb、2-3Kb和≥3Kb文库,数据产出比例为3:2:2。(例如:测8G的数据量,三个文库分别测3G、3G和2G,也可以根据不同物种调整不同文库的数据量);构建1-2Kb、2-3Kb、3-6Kb、≥6Kb四个文库(例如:测12G的数据量,四个文库分别测4G、4G,2G和2G。数据量分布一般是2:2:1:1或3:2:2:1。
注:根据甜菜三代全长转录组文献中报道还进一步验证了一个常识,多数原本转录本3'UTR+5'UTR长度>1Kb,所以一般不建议构建<1Kb文库,但研究目的是为了获得较为全面的转录本时才会建议构建<1Kb或0.5-1Kb文库。
9. 三代全长转录组建库测序的流程是什么?
图3. 三代转录组建库测序流程简图
简述以上流程:
a.全长cDNA合成:使用Clontech SMARTer PCR cDNA Synthesis Kit合成全长的cDNA;
b.片段选择及PCR扩增:采用BluePippinTM仪器直接进行片段筛选并进行扩增;
c.SMRTbell文库制备:将不同插入片段cDNA加上SMRTbell接头,并完成文库构建;
d.测序:文库进行质控后上机三代平台PacBio测序。
10. 三代全长转录组测序的生物信息分析流程是什么?具体有哪些分析内容?
图4. 三代全长转录组生物信息分析流程简图
表3. 三代全长转录组信息分析内容
有参考基因组物种
无参考基因组物种
(1) 原始数据处理及过滤;
(2) 测序数据质量评估;
(3)全长转录本判定;
(4)转录本聚类校正;
(5)与参考基因组序列比对;
(6)全长转录本比对注释;
(7)基因结构优化;
(8)可变剪接鉴定;
(9)新基因预测及CDS预测
(10)LncRNA预测
(11)基因融合鉴定
(1)原始数据处理及过滤;
(2)测序数据质量评估;
(3)全长转录本判定;
(4)转录本聚类与校正;
(5)全长转录本比对注释;
(6)预测编码蛋白框(CDS);
(7) SSR预测;
(8) LncRNA预测;
11. 三代全长转录组测序获得的Clean Reads中,全长序列所占比例是多少?
全长序列所占比例与测序量和建库长度以及表达量有关。没有准确的标准,一般全长比例会占到50%左右(与目前文献报道及官网测试数据水平一致)。
pacbio 三代全长转录组数据分析流程 Iso-Seq 3
Iso-Seq 建库
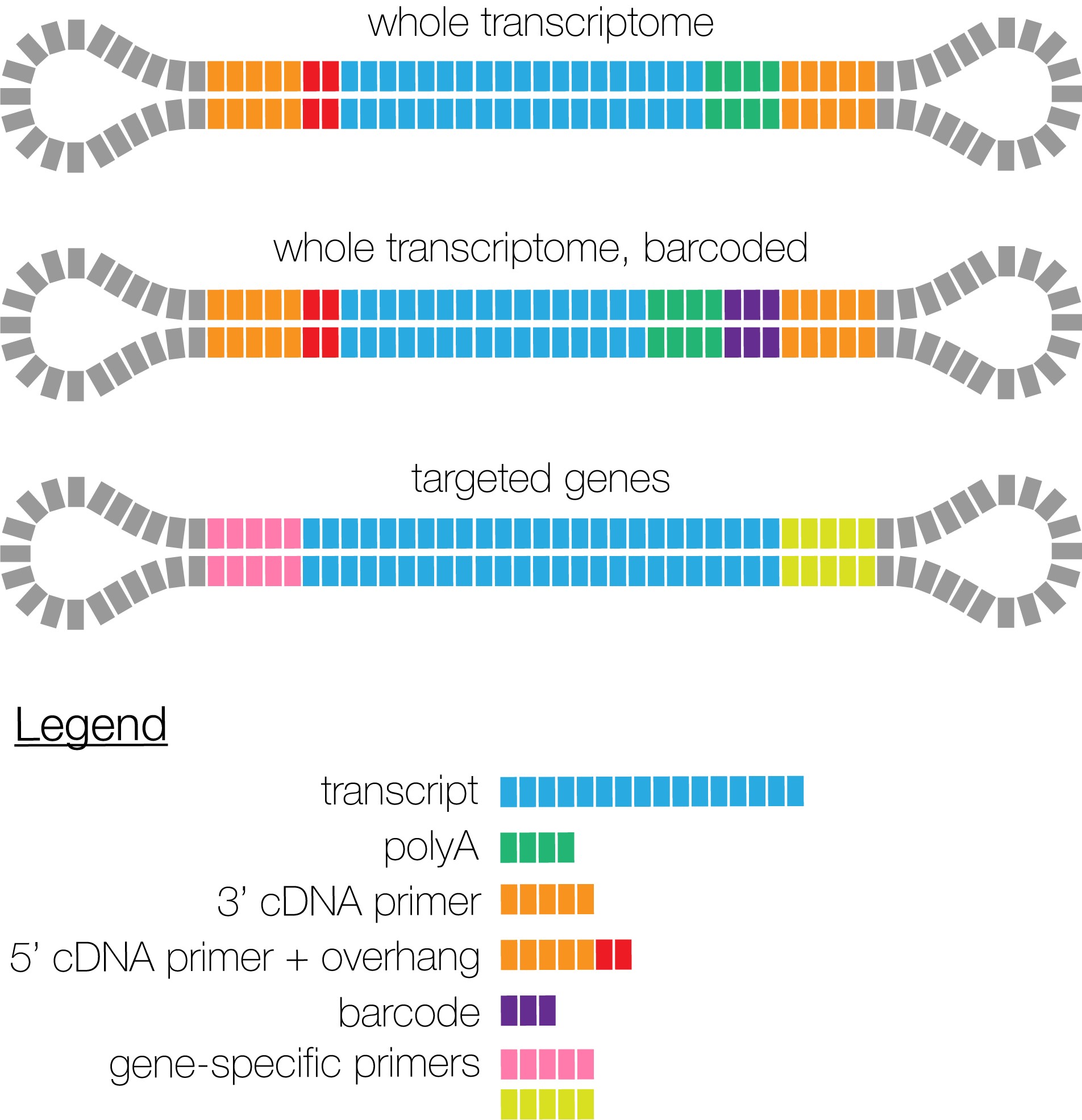
Iso-Seq的建库方案有如下三类:
- 整个库都是一个样品的全长转录组,不需要加barcode区分样品
- 不同样品的全长转录组,加上不同barcode ,可以放在一起进行建库测序
- 一些靶向获得的部分基因也可以进行全长转录组的测序
Iso-Seq 3进行数据分析
Iso-Seq3 进行全长转录组的分析,运行流程如下图所示:
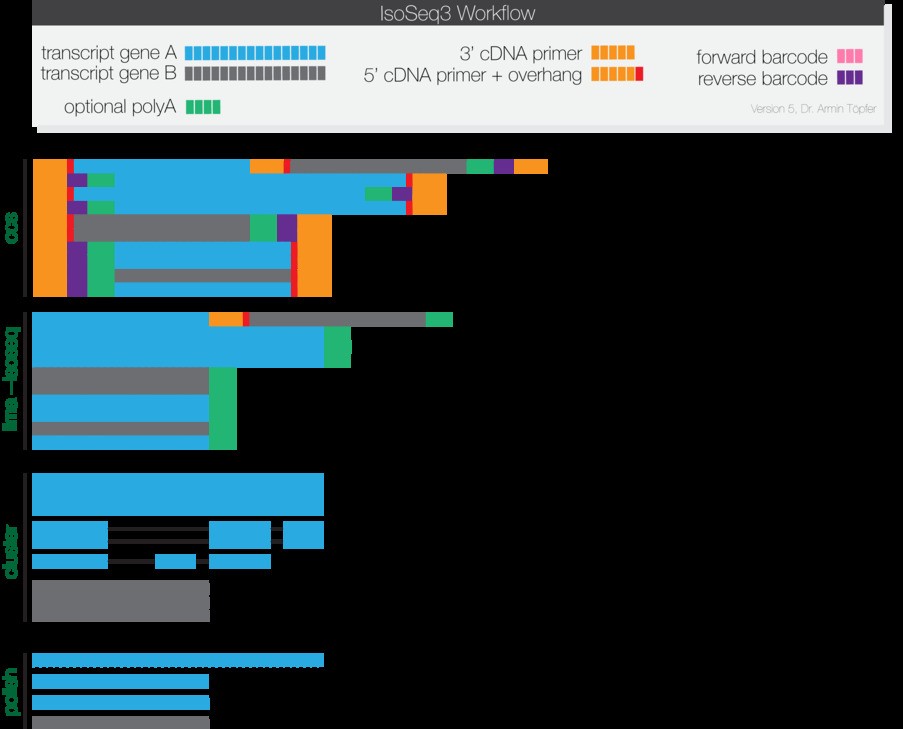
1 ccs(Circular Consensus Calling)
ccs 获取一致性的序列,要求每一条测序的reads至少有一端含有引物序列。
ccs test.subreads.bam ccs.bam --noPolish --minPasses 1
2 测序引物和barcode的去除
这一步是采用lima 来完成的,这个软件也是官方最新开发出来的程序,速度和准确度都较以往的算法有了很大的提升。还能够基于barcode序列区分不同的样品,
lima ccs.bam barcoded_primers.fasta demux.ccs.bam --isoseq --no-pbi
3 聚类(cluster)
聚类采用IsoSeq3 软件来完成,这一步会首先将ployA尾巴去除掉,并将连环结构去除掉,再对相似的序列进行聚类,最好形成全长的reads。
isoseq3 cluster demux.primer_5p--primer_3p.bam unpolished.bam --verbose
4 抛光(polish)
这一步主要是将聚类的转录本,合并成一个完整的一致性序列。
isoseq3 polish -j 16 unpolished.bam test.subreads.bam polished.bam
三代测序技术概述:
- PacBio和Oxford Nanopore测序的原理
- 三代测序的特点和应用
- 三代测序在转录组研究的优势和案例分享

三代测序基本分析流程


- 原始测序序列去除接头和错误序列
- 提取环形一致序列读长(CCS reads)
- CCS reads分类(包括全长和非全长CCS reads)
- CCS reads聚类(根据CCS reads序列的相似性)获得最终的转录本集合
- 最终转录本比对回基因组
- 转录本定量和可变剪接分析
PacBio测序平台基于其独特的单分子实时测序技术(Single Molecule Real Time,SMRT),通过其超长读长,均一的覆盖度,高度的一致性准及确性提供无与伦比的遗传信息深度解析。
用 GMAP/GSNAP软件进行RNA-seq的alignment
首先需要参考基因组:虽然软件本身提供了一个hg19的参考基因组,并且已经索引好了Human genome, version hg19 (5.5 GB)(http://research-pub.gene.com/gmap/genomes/hg19.tar.gz) ,但是下载很慢,而且不是对所有版本的GSNAP都适用。所以我这里对我自己的参考基因组进行索引。
软件安装
下面是我源码安装的代码
wget http://research-pub.gene.com/gmap/src/gmap-gsnap-2018-07-04.tar.gz
tar xf gmap-gsnap-2018-07-04.tar.gz
cd gmap-2018-07-04/
./configure --prefix=$HOME/opt/biosoft/gmap
make -j 20
软件使用
如下步骤假设你有一个物种的基因组序列和对应的CDS序列,分别命名为"reference.fa"和"cds.fa"
第一步:构建GMAP/GSNAP索引数据库
GMAP/GSNAP对FASTA文件中每个记录下的序列的长度有一定限制, 每一条不能超过4G, 能应付的了大部分物种了。
构建索引分为两种情况考虑,第一种是一个fasta文件包含所有的序列
~/opt/biosoft/gmap/bin/gmap_build -d reference reference.fa
第二种则是每个染色体的序列都单独存放在一个文件夹里,比如说你下载人类参考基因组序列解压后发现有N多个fasta文件, 然后你就想用其中几条染色体构建索引
~/opt/biosoft/gmap/bin/gmap_build -d reference Chr1.fa Chr2.fa Chr3.fa ...
注: 这里的-d表示数据K库的名字,默认把索引存放在gmap安装路径下的share里,可以用-D更改.此外还有一个参数-k用于设置K-mer的长度, 默认是15, 理论上只有大于4GB基因组才会有两条一摸一样的15bp序列(当然是完全随机情况下)。
第二步:正式比对
建立完索引之后就可以将已有的CDS或者EST序列和参考基因组序列进行比较。
~/opt/biosoft/gmap/bin/gmap -t 10 -d reference -f gff3_gene cds.fa > cds_gene.gff3
其中-t设置线程数, -d表示参考基因组数据库的名字, 都是常规参数。我比较感兴趣的参数是如何将序列输出成GFF格式. GMAP允许多种格式的输出,比如说-S只看联配的总体情况,而-A会显示每个比对上序列的联配情况, 还可以输出蛋白序列(-P)或者是genomic序列(-E). 但是做结构注释要的gff文件,参数就是-f gff3_gene, -f gff3_match_cdna, -f gff3_match_est。
参考文献
要想对一个软件有更好的认识,最好还是看看他们文章是怎么说的。
- GMAP: a genomic mapping and alignment program for mRNA and EST sequences
Bioinformatics 2005 21:1859-1875 Abstract Full Text, Thomas D. Wu and Colin K. Watanabe - Fast and SNP-tolerant detection of complex variants and splicing in short reads
Bioinformatics 2010 26:873-881 AbstractFull Text, Thomas D. Wu and Serban Nacu
PLEK:转录本蛋白编码潜能预测工具
在之前的文章中,我们介绍过CPC和CNCI这两款软件,可以用于预测lncRNA序列。其中CPC基于序列比对的方式,对于注释信息相对全面的物种分类效果较好,但是运行速度相对较慢,CNCI基于序列的三联体碱基组成来区分编码和非编码转录本,对于注释信息缺乏的物种,效果也不错,但是当序列中存在插入缺失时,其分类效果就变得很差。
在高通量测序产生的数据中,会存在一定的测序错误,虽然比例很低,但是基于这样的序列组装得到转录本然后去预测lncRNA, 对于CNCI这个软件而言,就会造成相当大的影响。
为了克服上述问题,需要一款运行速度又快,又可以一定程度上降低测序错误影响的lncRNA预测软件,PLEK软件就是基于这样的出发点进行开发的。PLEK软件通过序列的kmer构成来区分编码和非编码转录本,不需要通过比对来完成,所以运行速度较快,同时其性能受到测序错误的影响的概率较低,比较稳定。
可以看到PLEK的运行速度是最快的。该软件的源代码托管在sourceforge上,网址如下
https://sourceforge.net/projects/plek/files/
安装方式如下
wget https://sourceforge.net/projects/plek/files/PLEK.1.2.tar.gz
tar xzvf PLEK.1.2.tar.gz
cd PLEK.1.2
python PLEK_setup.py
基本用法如下
python PLEK.py
-fasta transcript.fa
-out output
-thread 10
只需要输入转录本对应的fasta格式的文件就可以了,输出文件output内容示意如下

第一列代表该转录本为coding还是non-coding, 第二列为打分值,打分值大于0为coding, 小于零为non-coding, 第三列为fasta文件中的序列标识符。
默认情况下会调用内置的svm模型,如果你有该物种已知的mRNA和lncRNA转录本序列,也可以构建自己的模型,代码如下
python PLEKModelling.py
-mRNA mRNAs.fa
-lncRNA lncRNAs.fa
-prefix 20190129
运行成功后,会生成后缀为.model和.range的两个文件。在预测时可以通过参数指定svm模型,用法如下
python PLEK.py
-fasta transcript.fa
-out output
-model 20190129.model
-range 20190129.range
-thread 10
来源:
https://www.cnblogs.com/wangprince2017/p/10852391.html
https://www.omicsclass.com/article/344
http://blog.sina.com.cn/s/blog_182e021ed0102xbqz.html
https://www.jianshu.com/p/3f331861c364