作业要求:
使用R语言,载入表达矩阵,然后设置好分组信息,统一用DEseq2进行差异分析,当然也可以走走edgeR或者limma的voom流程。
基本任务是得到差异分析结果,进阶任务是比较多个差异分析结果的异同点。
【1】安装DESeq2
1 # 下面是在R语言中操作 2 # 载入安装工具 3 > source("http://bioconductor.org/biocLite.R") 4 # 安装包 5 > biocLite("DESeq2") 6 # 载入包 7 > library("DESeq2")
DESeq2对于输入数据的要求:
1.DEseq2要求输入数据是由整数组成的矩阵。
2.DESeq2要求矩阵是没有标准化的。
【2】DESeq2进行差异表达分析
DESeq2分析差异表达基因简单来说只有三步:构建dds矩阵,标准化,以及进行差异分析。
# dds <- DESeqDataSetFromMatrix(countData = cts, colData = coldata, design= ~ batch + condition) #~在R里面用于构建公式对象,~左边为因变量,右边为自变量。
# dds <- DESeq(dds) #标准化
# res <- results(dds, contrast=c("condition","treated","control")) #差异分析结果
【3】构建dds矩阵
1 > library(DESeq2) # 加载包 2 > countData <- raw_count_filter[2:7] # 中括号中的数量要与condition中数量一致 3 > condition <- factor(c("control","control","control","hypoxia","hypoxia","hypoxia")) 4 > colData <- data.frame(row.names=colnames(countData),condition) 5 # raw_count_filter:是所有样品的count按照gene id融合后生成的矩阵。行为各个基因,列为各个样品,中间为计算reads。
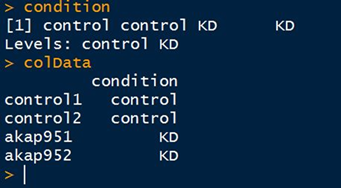
1 # 正式构建dds矩阵 2 > dds <- DESeqDataSetFromMatrix(countData,DataFrame(condition),design=~condition) 3 # 注意,condition前面是波浪线 4 > head(dds) # 查看一下构建好的矩阵
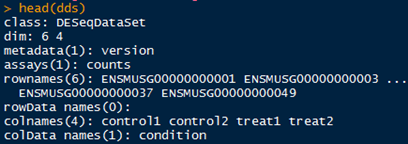
【4】对原始dds进行标准化
1 > dds2 <- DESeq(dds) # 对原始dds进行normalize
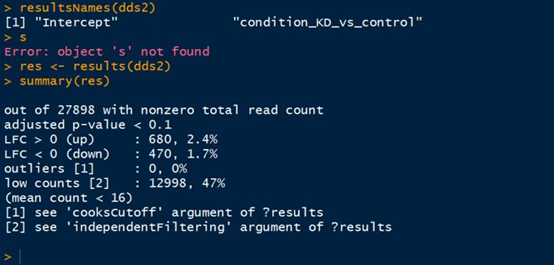
1 # 查看结果的名称,本次实验中是“intercept”,”condition_akap95_vs_control” 2 > resultsNames(dds2) 3 # 将结果用results()函数来获取,赋值给res变量 4 > res <- results(dds2) 5 # summary一下,看一下结果的概要信息 6 > summary(res)
【5】提取差异分析结果
1 # 获取padj(p值经过多重校验校正后的值)小于0.05,log2FoldChange的绝对值大于1的差异表达基因。 2 > table(res$padj<0.05) # 取p值小于0.05的结果 3 > res <- res[order(res$padj).] 4 > diff_gene_deseq2 <- subset(res,padj<0.05 & (log2FoldChange > 1 | log2FoldChange < -1)) 5 > diff_gene_deseq2 <- row.names(diff_gene_deseq2) 6 > resdata <- merge(as.data.frame(res),as.data.frame(counts(dds2,normalize=TRUE)),by="row.names",sort=FALSE) 7 # 将差异表达分析结果输出到csv文件 8 > write.csv(resdata,"d:/cs_file/r_file/diff_gene_control_vs_hypoxia.csv",row.names=FALSE)
补充:
用edgeR进行基因差异表达分析
1 # 加载edgeR包 2 library(edgeR) 3 ##跟DESeq2一样,导入数据,预处理(用了cpm函数) 4 exprSet<- read.table(file = "mouse_all_count.txt", sep = " ", header = TRUE, row.names = 1, stringsAsFactors = FALSE) 5 group_list <- factor(c(rep("control",2),rep("Akap95",2))) 6 exprSet <- exprSet[rowSums(cpm(exprSet) > 1) >= 2,] 7 8 ##设置分组信息,并做TMM标准化 9 exprSet <- DGEList(counts = exprSet, group = group_list) 10 exprSet <- calcNormFactors(exprSet) 11 12 ##使用qCML(quantile-adjusted conditional maximum likelihood)估计离散度(只针对单因素实验设计) 13 exprSet <- estimateCommonDisp(exprSet) 14 exprSet <- estimateTagwiseDisp(exprSet) 15 16 ##寻找差异gene(这里的exactTest函数还是基于qCML并且只针对单因素实验设计),然后按照阈值进行筛选即可 17 et <- exactTest(exprSet) 18 tTag <- topTags(et, n=nrow(exprSet)) 19 diff_gene_edgeR <- subset(tTag$table, FDR < 0.05 & (logFC > 1 | logFC < -1)) 20 diff_gene_edgeR <- row.names(diff_gene_edgeR) 21 write.csv(tTag$table,file = "control_vs_Akap95_edgeR.csv")
相关理论知识
构建dds矩阵需要:
表达矩阵 即上述代码中的countData,就是我们前面通过read count计算后并融合生成的矩阵,行为各个基因,列为各个样品,中间为计算reads或者fragment得到的整数。我们后面要用的是这个文件(mouse_all_count.txt)
样品信息矩阵即上述代码中的colData,它的类型是一个dataframe(数据框),第一列是样品名称,第二列是样品的处理情况(对照还是处理等),即condition,condition的类型是一个factor。这些信息可以从mouse_all_count.txt中导出,也可以自己单独建立。
差异比较矩阵 即上述代码中的design。 差异比较矩阵就是告诉差异分析函数是要从要分析哪些变量间的差异,简单说就是说明哪些是对照哪些是处理。
