1、eQTL、mQTL共定位分析的作用
eQTL、mQTL共定位分析属于Post-GWAS的一项重要工作,旨在GWAS结果的基础上鉴定与表型相关的eQTL和mQTL位点。
传统的GWAS是将全基因组范围内的常见变异进行关联分析,鉴定与表型相关的基因座,但鉴定出来的位点大多数位于基因间隔区,其如何通过基因或者通路影响表型很难被阐述。
基于此,开发了eQTL、mQTL共定位分析方法。
其原理是利用已有数据库公布的eQTL、mQTL位点,结合GWAS summary数据,鉴定与表型相关的eQTL和mQTL位点。
2、eQTL共定位分析
下载安装SMR:
wget https://cnsgenomics.com/software/smr/download/smr_Linux.zip
unzip smr_Linux.zip
下载eQTL数据(注意我这里下载的是hg19版本的,如果你的GWAS数据不是hg19,请自行更改对应的基因组版本的数据):
数据来源:https://cnsgenomics.com/software/smr/#eQTLsummarydata
wget https://cnsgenomics.com/data/SMR/westra_eqtl_hg19.zip
unzip westra_eqtl_hg19.zip
wget https://cnsgenomics.com/data/SMR/cage_eqtl_data_lite_hg19.tar.gz
tar -zxvf cage_eqtl_data_lite_hg19.tar.gz
wget https://cnsgenomics.com/data/SMR/GTEx_V7_cis_eqtl_summary.tar.gz
tar -zxvf GTEx_V7_cis_eqtl_summary.tar.gz
eQTL共定位分析(以westra_eqtl_hg19数据为例):
smr_Linux --bfile file --gwas-summary mygwas.ma --beqtl-summary westra_eqtl_hg19 --out mygwas --thread-num 10
参数解读:
file为PLINK的二进制格式文件,不了解的话请见链接https://www.cnblogs.com/chenwenyan/p/6095531.html
mygwas.ma为GWAS的summary文件,包含SNP、A1、A2、freq、b、se、p、N,N指样本数(N没有的话,可以直接赋予NA),内容如下所示:
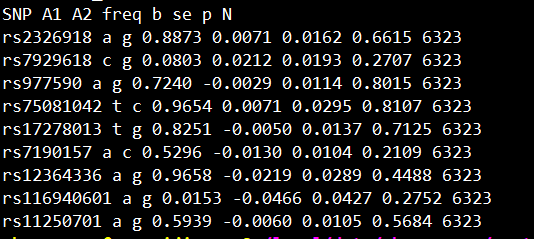
westra_eqtl_hg19为eQTL数据,该数据通过westra_eqtl_hg19.zip解压得到
mygwas为生成的共定位结果文件名
eQTL共定位结果(以westra_eqtl_hg19数据为例):
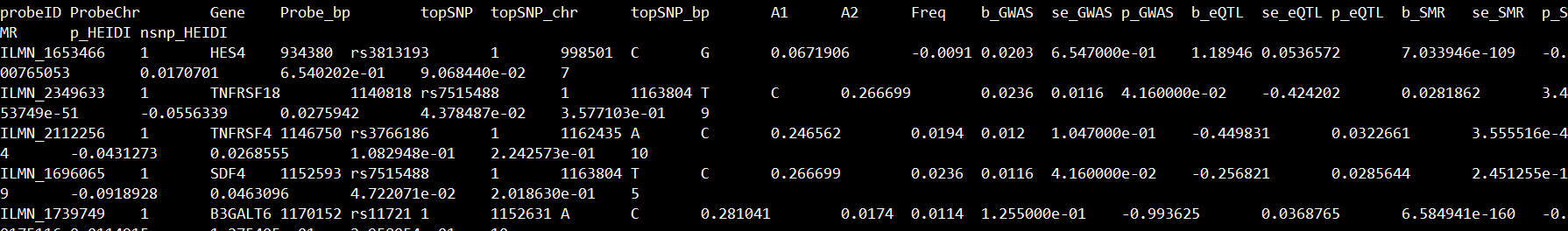
每一列代表的意思:Columns are probe ID, probe chromosome, gene name, probe position, tans-eQTL chromosome, left boundary of the trans-region, right boundary of the trans-region, SNP name, SNP chromosome, SNP position, the effect (coded) allele, the other allele, frequency of the effect allele (estimated from the reference samples), effect size from GWAS, SE from GWAS, p-value from GWAS, effect size from eQTL study, SE from eQTL study, p-value from eQTL study, effect size from SMR, SE from SMR, p-value from SMR, p-value from HEIDI test, and number of SNPs used in the HEIDI test.
3、mQTL共定位分析
下载mQTL数据(注意我这里下载的是hg19版本的,如果你的GWAS数据不是hg19,请自行更改对应的基因组版本的数据):
数据来源:https://cnsgenomics.com/software/smr/#mQTLsummarydata
wget https://cnsgenomics.com/data/SMR/LBC_BSGS_meta.tar.gz
tar -zxvf LBC_BSGS_meta.tar.gz
mQTL共定位分析(以LBC_BSGS_meta的1号染色体数据为例):
smr_Linux --bfile file --gwas-summary mygwas.ma --beqtl-summary /LBC_BSGS_meta/bl_mqtl_chr1 --out mygwas --thread-num 10
参数解读:
file和mygwas.ma与eQTL共定位分析的输入文件相同
/LBC_BSGS_meta/bl_mqtl_chr1为mQTL数据,该数据通过LBC_BSGS_meta.tar.gz解压得到
mygwas为生成的共定位结果文件名
mQTL共定位结果(以LBC_BSGS_meta数据为例):

每一列代表的意思同eQTL的结果分析
4、关于eQTL、mQTL共定位分析相关的文献推荐
文献题目:Summary-Based Methylome-Wide Association Analyses Suggest Potential Genetically Driven Epigenetic Heterogeneity of Alzheimer's Disease
不想看英文题目:基于摘要统计的甲基化全基因组分析揭示阿尔兹海默症的表观异质性
杂志和影响因子:J Clin Med (IF: 5.688)
分析方法:结合阿尔兹海默症遗传风险位点和公共数据库公开的全血、大脑的mQTL位点进行SMR分析,鉴定与阿尔兹海默症和DNA甲基化变化相关的SNP,使用GSA-SNP2包进行通路富集分析。
结论:找到152个探针、113个基因与阿尔兹海默症相关,其中10个基因均在血液和大脑甲基化中达到显著水平。将个体按照性别、有无高血压分组,分别找到22和79个显著探针位点与阿尔兹海默症相关,其可能是阿尔兹海默症异质性的原因。
文章链接:https://pubmed.ncbi.nlm.nih.gov/32429084/
文献题目: Multivariate genomic scan implicates novel loci and haem metabolism in human ageing
不想看英文题目: 多元基因组扫描揭示人类衰老中新的基因座和血红素代谢
杂志和影响因子: Nat Commun (IF: 11.878)
分析方法: 使用MANOVA对寿命、健康寿命、父母寿命三个不同的GWAS summary文件进行荟萃分析。随后对感兴趣的基因座按照性别和年龄进行分层分析,用GWAS catalog和PhenoScanner查找感兴趣基因的相关表型。使用SMR进行基因表达共定位分析,使用FUMA的Gene2Func进行基因富集分析,使用TwoSampleMR进行孟德尔随机化随机化;
结论: 通过荟萃分析,找到了10基因影响三个表型,其中5个(FOXO3,SLC4A7,LINC02513,ZW10和FGD6)是以前未报道过的。这10个基因座大多数与心血管疾病有关,其表达活性随年龄发生变化。有78个基因富集在衰老通路上,涉及的通路有DNA损伤,凋亡和体内平衡,血红素代谢。
文章链接:
https://pubmed.ncbi.nlm.nih.gov/32678081/
