1)知识简介
--------------------------------------------------------
1.1)测序质量值
首先在了解fastq,fasta之前,了解一下什么是质量值。phred软件在对reads进行base calling的时候会给出每一个碱基的质量值,这个质量值的计算与测序预期错误率相关(estimated probability of error):
Phred Quality Score Probability of incorrect base call Base call accuracy
10 1 in 10 90 %
20 1 in 100 99 %
30 1 in 1000 99.9 %
40 1 in 10000 99.99 %
50 1 in 100000 99.999 %
除此之外还有solexa标准,即将p换成了p/(1-p),其他完全按照sanger的定义来做。当测序质量很高的情况下两种形式几乎没区别,但低质量的碱基则有区别了(见下图)

Qscore与p之间的关系,其中红线表示Q=-10 log10p标准,黑色实线表示Q=-10 log10p/(1-p)标准。
1.2)ACII码
为了方便储存及可读这些信息,利用可打印的ACII码将这些质量值转化为单字符single characters (or bytes)。ASCII 字符集,最基本的包含了128 个字符。其中前 32 个, 0-31 ,即 0x00-0x1F ,都是不可见字符,这些字符,为控制字符。可见字符为32–126。sanger-fastaq格式用 ASCII 33–126 来表示phred 质量值 0 到93 。举例来说:一般地,碱基质量从0-40,既ASCii码为从 “!”(0+33)到“I”(40+33)。如果某碱基测序出错的概率为0.001,则Q应该为30。则30+33=63,那么63对应的ASCii码为“?”,在第四行中该碱基对应的质量代表值即为“?”。

2)fastq格式
fastq格式是一个文本格式用于贮存生物学序列及其相应质量值(通常是核酸序列的)。为了简介,这些序列以及质量信息使用ASCII字符标示。该格式最初由Sanger开发,目的是将FASTA序列与质量数据放到一起,目前已经成为高通量测序结果的事实标准。通常fastq文件中每一个序列含有4行信息(如下):
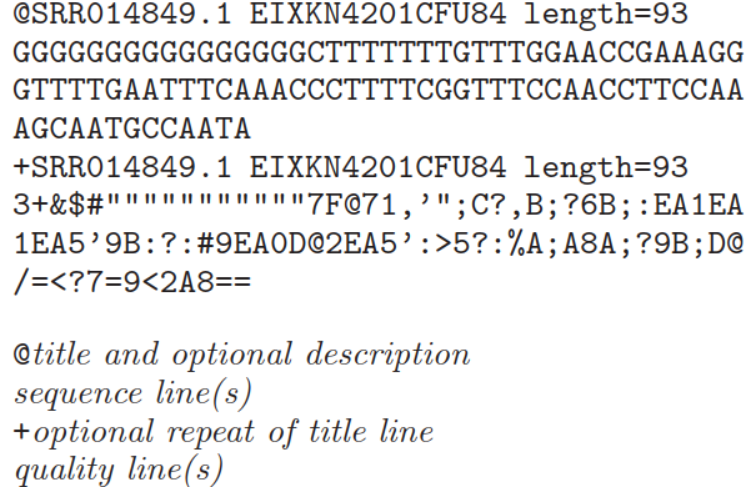
第一行:序列标识,以‘@’开头。格式比较自由,允许添加注释等相关的描述信息,描述信息以空格分开。如示图中描述信息加入了NCBI的另一个ID名称,及长度信息
第二行:表示序列信息,制表符或者空格不允许出现。一般是明确的DNA或者RNA字符,通常是大写,在一些文本文件中,小写或者大小写混杂或者含有gap符号是有特殊含义。
第三行:用于将测序序列和质量值内容分离开来。以‘+’开头,后面是描述信息等,或者什么也不加。如果“+”后面有内容,该内容与第一行“@”后的内容相同;
第四行:表示质量值,每个字符与第二行的碱基一一对应,按照一定规则转换为碱基质量得分,进而反映该碱基的错误率,因此字符数必须和第二行保持一致。对于每个碱基的质量编码标示,不同的软件采用不同的方案 。目前有5种:
1、Sanger,Phred quality score:值的范围从0到93,对应的ASCII码从33到126,但是对于测序数据(raw read data)质量得分通常小于60,序列拼接或者mapping可能用到更大的分数。
2、Solexa/Illumina 1.0, Solexa/Illumina quality score:值的范围从-5到62,对应的ASCII码从59到126,对于测序数据,得分一般在-5到40之间;
3、Illumina 1.3+,Phred quality score:值的范围从0到62对应的ASCII码从64到126,低于测序数据,得分在0到40之间;
4、Illumina 1.5+,Phred quality score:但是0到2作为另外的标示,详见http://solexaqa.sourceforge.net/questions.htm#illumina
5、Illumina 1.8+
不同的标准之间可以相互转化换,感兴趣可以自己查资料,这里不做详细介绍。注意:第二行@字符,第三行+字符,在第四行质量值中会出现,有时也会在行首出现,因此在处理fastq格式的时候要格外的关注。
3)fasta格式
------------------------------------
3.1)fasta格式最初来自FASTA软件包,也是一种文本格式,以单字符( single-letter codes)贮存核酸或者蛋白序列信息,允许在序列前加注释信息。由2部分信息组成:
>gi|5524211|gb|AAD44166.1| cytochrome b [Elephas maximus maximus]
LCLYTHIGRNIYYGSYLYSETWNTGIMLLLITMATAFMGYVLPWGQMSFWGATVITNLFSAIPYIGTNLVEWIWGGFSVDKATLNRFFAFHFILPFTMVALAGV
HLTFLHETGSNNPLGLTSDSDKIPFHPYYTIKDFLGLLILILLLLLLALLSPDMLGDPDNHMPADPLNTPLHIKPEWYFLFAYAILRSVPNKLGGVLALFLSIV
IGLMPFLHTSKHRSMMLRPLSQALFWTLTMDLLTLTWIGSQPVEYPYTIIGQMASILYFSIILAFLPIAGXIENY
第一部分:以>号开始,紧接着序列的标识符 ,注意区分大小写,且不能出现空格,空格表示序列标识符结束; 随后是序列的描述信息。
第二部分:以序列本身信息,使用既定的核苷酸或氨基酸编码符号,大小写都可以。直到遇到下一个>结束。所有来源于NCBI的序列都有一个gi号“gi|gi_identifier”,gi号由数字组成,具有唯一性。一条核酸或者蛋白质改变了,将赋予一个新的gi号(这时序列的接收号可能不变)。gi号后面是序列的标识符,标识符由序列来源标识、序列标识(如接收号、名称等)等几部分组成,他们之间用“|”隔开,如果某项缺失,可以留空但是“|”不能省略。
3.2)fasta格式在拓展的文件命名中,一般会约定俗成,具体见下表格:
| Meaning | Notes | |
|---|---|---|
| fasta | generic fasta | Any generic fasta file. Other extensions can be fas, fa, seq, fsa |
| fna | fasta nucleic acid | Used generically to specify nucleic acids. |
| ffn | FASTA nucleotide of gene regions | Contains coding regions for a genome. |
| faa | fasta amino acid | Contains amino acids. A multiple protein fasta file can have the more specific extension mpfa. |
| frn | FASTA non-coding RNA | Contains non-coding RNA regions for a genome, in DNA alphabet e.g. tRNA, rRNA |
4)习题(fq练习文件已boweti2中的示例文件reads_1.fq)
--------------------------------------------------------------------------------------------------
4.1) fq文件中的质量值是如何产生的?
4.2) fq的质量在转化成ACII码的时候,为什么不选择前32个?(0-32)
4.3) fq的质量值在转化成ACII码的时候,为什么不从32开始,而是从33开始?
4.4)在统计fq有多少条序列的时候能不能直接grep '@' read_1.fq | wc -l ?为什么?
4.5)在sanger和solexa标准中,测序的错误率与质量值之间的差别在哪里?
4.6)fasta格式起源于什么地方?4.7)fasta文件的命名有没有特殊的含义?
4.8)fasta序列标识符是如何对应来自不同的数据库来源的?
4.9)转换fasta与fasta的软件有哪些?
4.10)fasta中的字母大小写有没有特殊的含义?
4.11) 统计reads_1.fq文件中共有多少条序列信息
4.12)输出所有的reads_1.fq文件中的标识符(即以@开头的那一行)
4.13)计算reads_1.fq 所有的reads中N的总数
4.14)统计reads_1.fq 中测序碱基为Q30的含量
4.15)统计reads_1.fq 中测序碱基质量为所有大于Q20的碱基含量
4.16)将reads_1.fq转为reads_1.fa文件(即将fastq转化为fasta)
4.17)计算reads_1.fa文件中GC数量
4.18)统计文件中reads_1.fa碱基总数
4.19)计算reads_1.fa文件中GC含量百分比
4.20)过滤掉reads_1.fa文件中N含量超过10%的reads,并统计有多少条
5) 参考资源
--------------------------------------------------------------
The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants
https://en.wikipedia.org/wiki/FASTQ_format
https://en.wikipedia.org/wiki/FASTA
http://boyun.sh.cn/bio/?p=1901
https://blog.csdn.net/open2open2/article/details/26706969