下面的图来自文章 https://www.sciencedirect.com/science/article/pii/S0092867415005498
Droplet Barcoding for Single-Cell Transcriptomics Applied to Embryonic Stem Cells
Cell, Volume 161, Issue 5, 21 May 2015, Pages 1187-1201
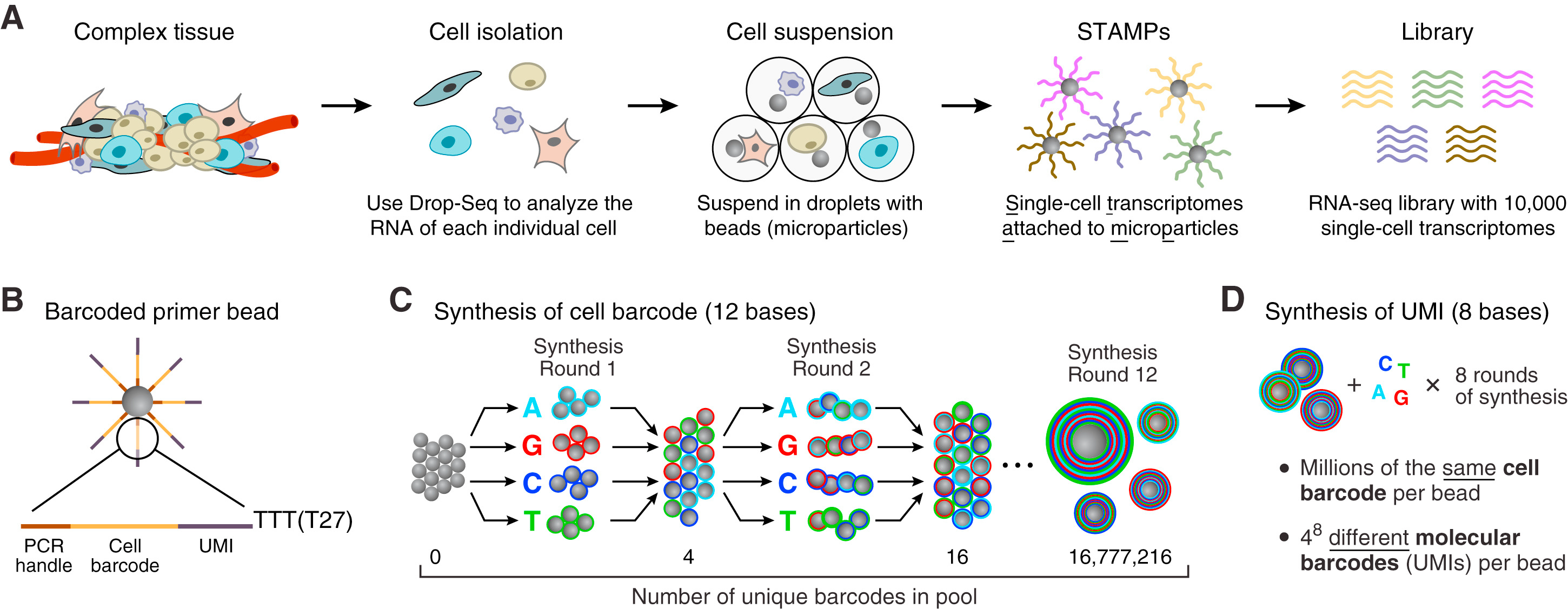
图一
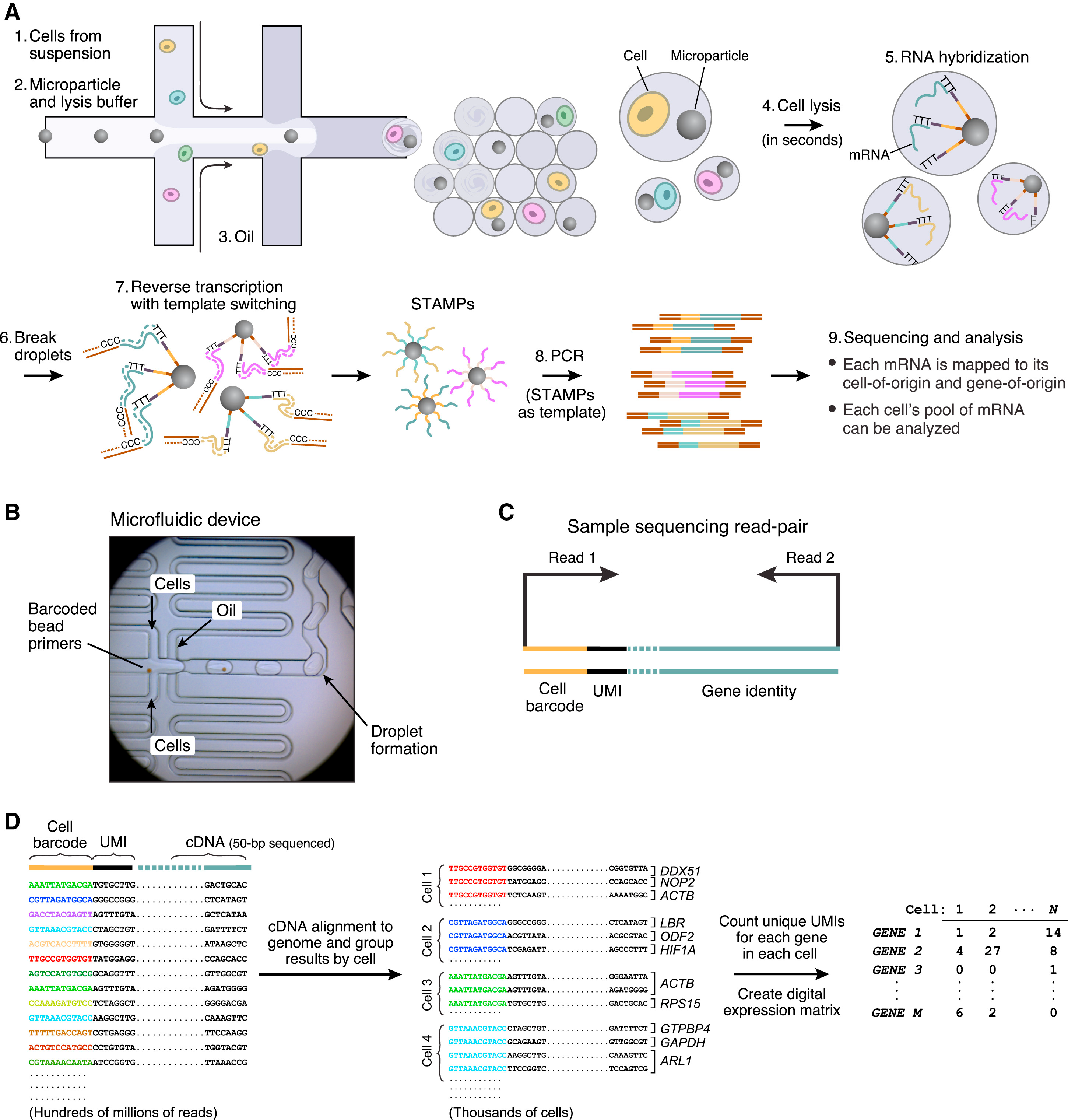
图二
首先,为了保证细胞在标记的过程中是分开的,发明了一个叫做微流体设备的东西(microfuidic device)
里面有三个上样孔,在其中可以分别加入
里面有三个上样孔,在其中可以分别加入
1、样本细胞悬液,
2、Gel Beads
3、partitioning oil。
像下图中一样。

然后,它就可以启动图二A中的流程,当细胞流过时,会被与beads结合然后被油滴包裹,形成一个油包水的密闭小液滴(droplet)。细胞和microparticle(beads)相遇会裂解,释放出里面的各种物质,RNA(mRNA,tRNA,rRNA),蛋白质,脂质,DNA等等各种物质。因为beads上联接了接头,其中有一段是ploy (dT)序列(30)个,在细胞裂解释放的核酸中,只有mRNA带有polyA的尾巴,于是这段beads上面ploy (dT)就可以从众多的裂解产物里捕获到mRNA。这也就是Russell问我为什么Drop-seq要用3'端测序的原因了。
Maxter Mix中带有反转录试剂,当mRNA被捕获后,就可以从它的3‘端开始作为模板,进行反转录出cDNA的第一条链,这第一条链就沿着poly(dT)序列延申,长在了beads上,形成了图二7中的STAMPs,接着我们把序列洗脱,以cDNA的第一条链为模板,进行PCR,合成cDNA的第二条链,然后就是我们熟悉的cDNA扩增,illumina测序。
Maxter Mix中带有反转录试剂,当mRNA被捕获后,就可以从它的3‘端开始作为模板,进行反转录出cDNA的第一条链,这第一条链就沿着poly(dT)序列延申,长在了beads上,形成了图二7中的STAMPs,接着我们把序列洗脱,以cDNA的第一条链为模板,进行PCR,合成cDNA的第二条链,然后就是我们熟悉的cDNA扩增,illumina测序。
我们如何知道那些序列是来自那个细胞的?或者说,single
cell的RNA-seq和bulk的RNA-seq的最大区别在哪里呢?是Barcode,或者说是Cell
barcode。每一个beads上都有着相同的Cell barcode,而beads与beads间的Cell
barcode是不同的,假设每个beads只捕获一个cell,那么则每个cell都被cell barcode 单独标记了。
如何保证每一beads只捕获一个cell呢?第一是控制cell和beads的流速,第二是beads的数目远远超过cell的数目,即绝大多数的beads都是空的,只有少数的才捕获到了cell。但是还是有个别的droplet里面会两个或者更多的细胞,这就需要我们去QC啦。
single cell RNA-seq的barcode这么神奇,我们结合10x Genomics的说明书一起来分步骤详解一下。
如何保证每一beads只捕获一个cell呢?第一是控制cell和beads的流速,第二是beads的数目远远超过cell的数目,即绝大多数的beads都是空的,只有少数的才捕获到了cell。但是还是有个别的droplet里面会两个或者更多的细胞,这就需要我们去QC啦。
single cell RNA-seq的barcode这么神奇,我们结合10x Genomics的说明书一起来分步骤详解一下。
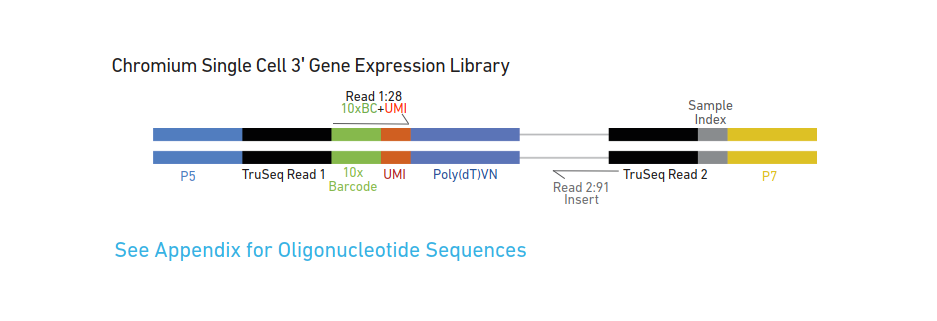
先来看看这些区段都是用来做什么的。
由内向外:
Poly(dT):用来和polyA结合,捕获mRNA
UMI:用来标记不同的PCR产物(数count)
10xBarcode:用来标记不同的细胞
Sample Index:用来标记不同的样本
Truseq Read 1、2 :用来进行连接beads,cDNA的PCR扩增和加P7接头
P5和P7:用来进行illumina的桥式PCR测序。
在这些序列中,P5、P7、Read 1、2 的序列是已知的
和beads上连接的序列最开始只有Read1 ,10x Barcode,和poly(dT),其他的这些接头是怎样一步步加上,并且发挥作用的?10xGenomics的说明书上给了具体步骤。我看下来,觉得最主要的思想,就是用已知的序列设计引物,PCR出未知的序列。
由内向外:
Poly(dT):用来和polyA结合,捕获mRNA
UMI:用来标记不同的PCR产物(数count)
10xBarcode:用来标记不同的细胞
Sample Index:用来标记不同的样本
Truseq Read 1、2 :用来进行连接beads,cDNA的PCR扩增和加P7接头
P5和P7:用来进行illumina的桥式PCR测序。
在这些序列中,P5、P7、Read 1、2 的序列是已知的
和beads上连接的序列最开始只有Read1 ,10x Barcode,和poly(dT),其他的这些接头是怎样一步步加上,并且发挥作用的?10xGenomics的说明书上给了具体步骤。我看下来,觉得最主要的思想,就是用已知的序列设计引物,PCR出未知的序列。
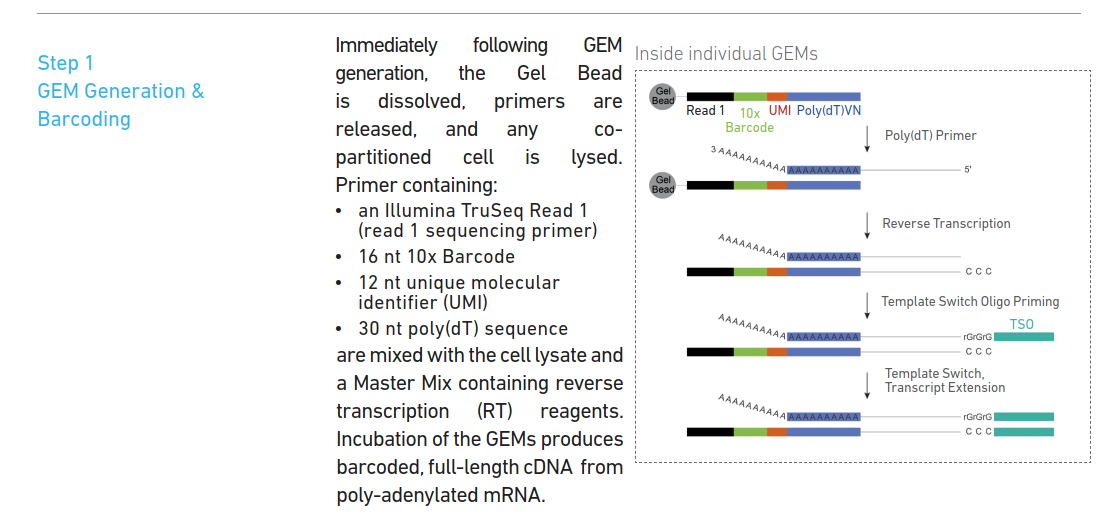
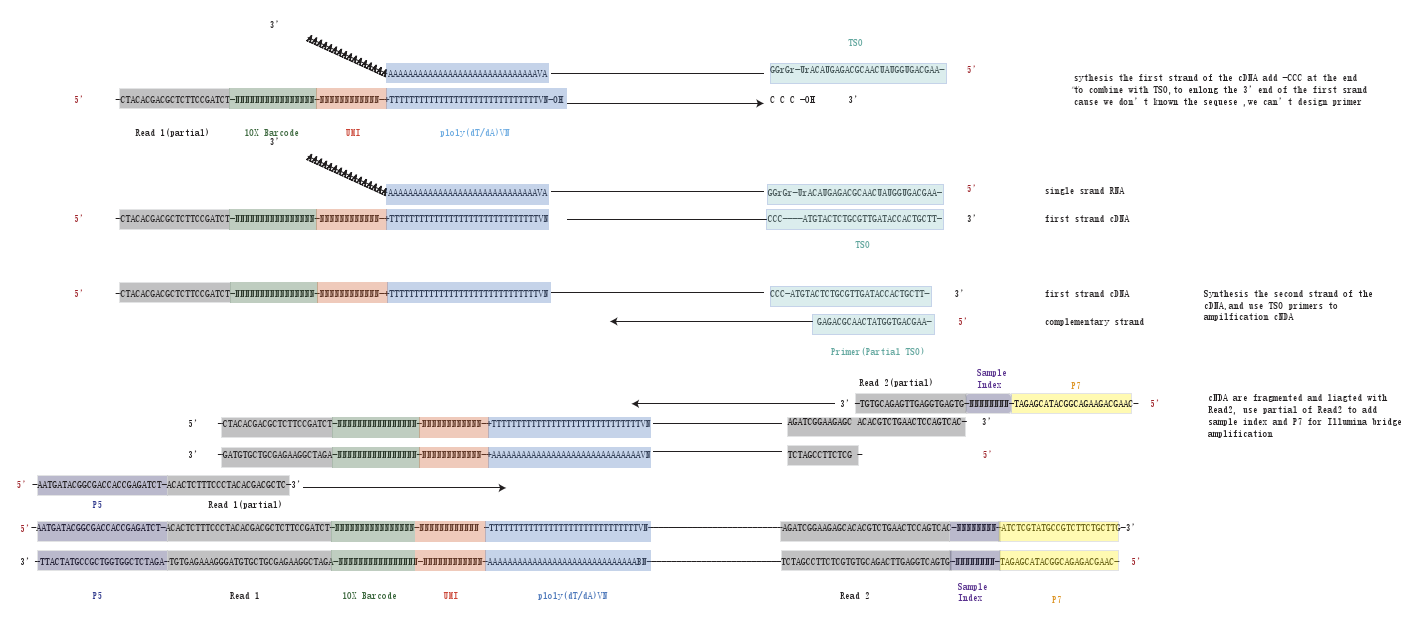
利用Poly(dT)来捕获mRNA,反转录出cDNA的第一条链,并且在cDNA的末端加上CCC序列,和TSO序列的GGG序列互补,并以TSO序列为模板,继续延申。这其实很重要,因为中间cDNA的序列我们是不知道的,如果不加上这个接头,就没有办法设计引物,合成cDNA的第二条链。

对TSO序列和Read1 设计引物,合成cDNA的第二条链,并完成cDNA的扩增。

因为II代测序illumina测序不能测很长,约为200-700bp,所以不能测得mRNA全长,所以会把合成的cDNA利用酶打断到illumina能测的长度,然后再两边加上接头,Read2。 设计部分与Read1和Read2重叠的引物,加上P5,P7和sample index,就可以去进行illumina测序了。
链接:https://www.jianshu.com/p/70dcddd2084e