摘要
甲基化在真核生物基因组序列中广泛存在,其中5mC最为普遍,在真核生物基因组中也有发现6mA。捕获基因组中的甲基化状态的常用技术是全基因组甲基化测序(WGBS)和简化甲基化测序(RRBS),而随着第三代测序技术的完善,ONT单分子纳米孔测序可以从单分子的角度来检出甲基化的胞嘧啶和腺嘌呤电流的变化,从而实现由基因组中的一段序列中检出5mC和6mA,然而精确地从单碱基级别检出5mC和6mA扔具有挑战。本文利用第三代ONT测序技术获得的序列及其电信号来检出真核生物全基因组范围的5mC和6mA甲基化状态。
背景
DNA甲基化主要发生在脱氧核糖核苷酸的第五位的胞嘧啶和第六位的腺嘌呤,前者普遍存在于真核生物,后者在原核生物中广泛存在,也有研究报道6mA存在于真核生物。这样的甲基化状态在ONT测序仪捕捉到的电流信号中,不仅单碱基的电流会发生改变,而且其上下文的一段基因组序列也会发生改变。基于此,一些生物信息学软件先后被开发出来针对于这两种甲基化的检出有各自的优缺点。有研究指出在真核基因组中检出5mC和6mA准确度较高的软件分别为nanopolish[1]和tombo[2]。
利用nanopolish检出真核生物基因组中5mC的甲基化位置
材料和方法
利用minION平台对目标生物血液提取的DNA不打断建库并进行全基因组测序,获得12G序列及其电信号文件。安装nanopolish(v0.13.2)。
步骤
- 建立索引
nanopolish index -d fast5_files/ output.fastq
- 比对
minimap2 -a -x map-ont reference.fasta output.fastq | samtools sort -T tmp -o output.sorted.bam
samtools index output.sorted.bam
- Calling methylation
nanopolish call-methylation --progress -q cpg -t NCPU --verbose -r reads.fastq -b output.sorted.bam -g reference_genome.fasta > nanopolish_call_methylation.tsv
- 筛选高置信度的甲基化和未甲基化位点
calculate_methylation_frequency.py[3]
nanopore-methylation-utilities/mtsv2bedGraph.py[4]
scripts/calculate_methylation_frequency.py -c 2 methylation_calls.tsv > methylation_frequency.tsv
# or
python nanopore-methylation-utilities/parseMethylbed.py frequency -i methylation_calls.tsv -o methylation_calls_freq.tsv --verbose -m cpg -u 2 -l -2
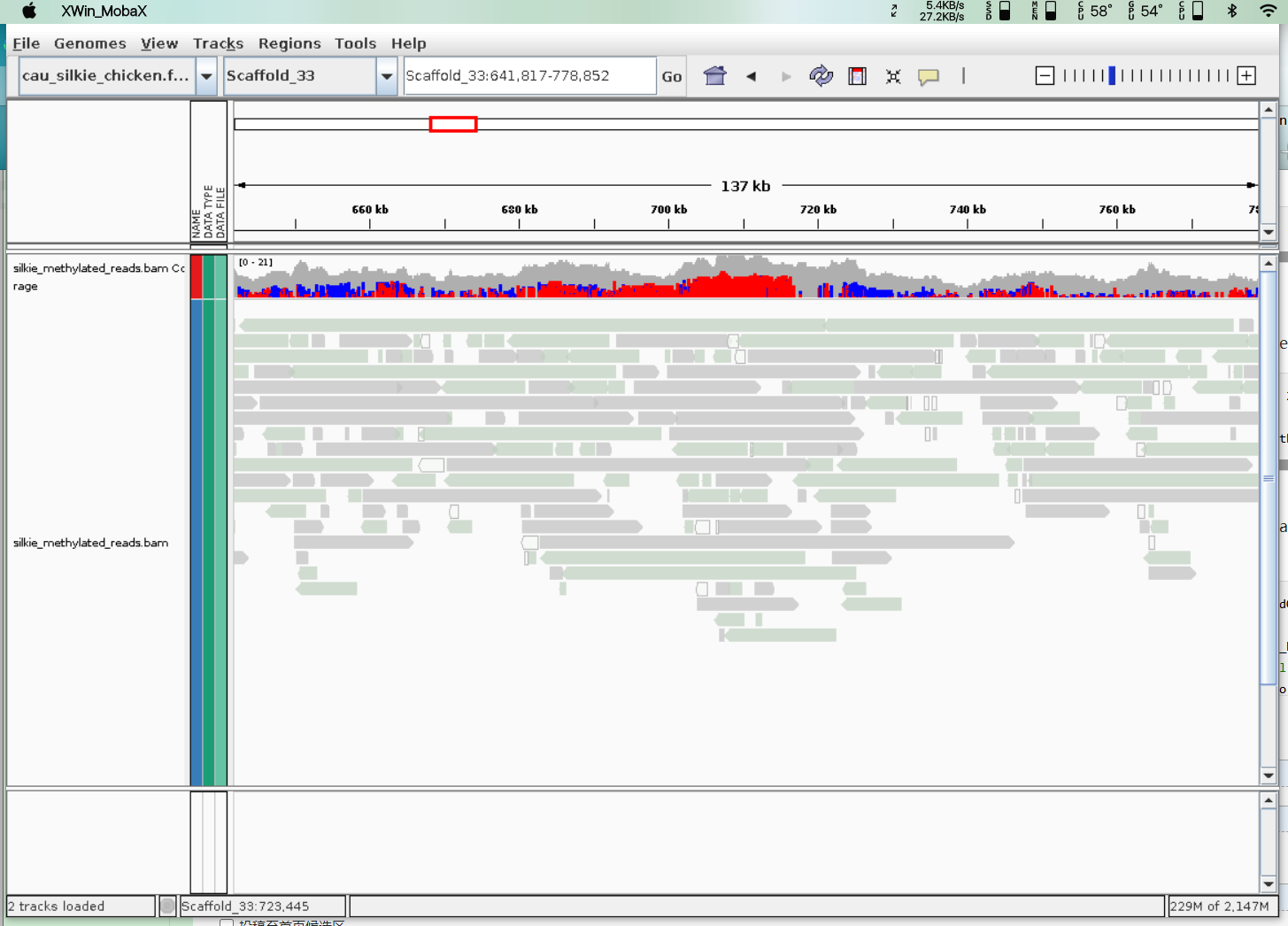
- IGV可视化或UCSC可视化
IGV可视化[5]
python nanopore-methylation-utilities/mtsv2bedGraph.py --verbose -c 2 -i methylation_calls.tsv -q cpg -g reference_genome.fasta | sort -k1,1 -k2,2n | bgzip > methylation_calls.bed.gz
tabix -p bed methylation_calls.bed.gz
python nanopore-methylation-utilities/convert_bam_for_methylation.py -t 100 --verbose --remove_poor -c methylation_calls.bed.gz -f reference_genome.fasta -b output.sorted.bam | samtools sort -o methylation_calls.bed.remove_no_or_poor_methylation_reads.bam
# if all reads' coverage was needed, remap all reads to reference to find out
samtools index methylation_calls.bed.remove_no_or_poor_methylation_reads.bam
# now bam file can be loaded to igv via their bisulfite mode to see methylation sites and unmethylation sites.