转:https://shengxin.ren/article/16
https://www.cnblogs.com/lmt921108/p/7442699.html
批量下载SRA http://www.360doc.com/content/18/0428/15/48272598_749456477.shtml
我的下载的数据在/home/username/ncbi/public/sra
SRA(Sequence ReadArchive)数据库是用于存储二代测序的原始数据,包括 454,Illumina,SOLiD,IonTorrent,Helicos 和 CompleteGenomics。除了原始序列数据外,SRA现在也存在raw reads在参考基因的比对信息。
根据SRA数据产生的特点,将SRA数据分为四类:
-
Studies-- 研究课题
-
Experiments-- 实验设计
-
Runs-- 测序结果集
-
Samples-- 样品信息
SRA中数据结构的层次关系为:Studies->Experiments->Samples->Runs.
-
Studies是就实验目标而言的,一个study 可能包含多个Experiment。
-
Experiments包含了Sample、DNA source、测序平台、数据处理等信息。
-
一个Experiment可能包含一个或多个runs。
-
Runs 表示测序仪运行所产生的reads。
SRA数据库用不同的前缀加以区分:
-
ERP或SRP表示Studies;
-
SRS 表示 Samples;
-
SRX 表示 Experiments;
-
SRR 表示 Runs;
使用:
搜索相关研究的疾病,选择相应数据集
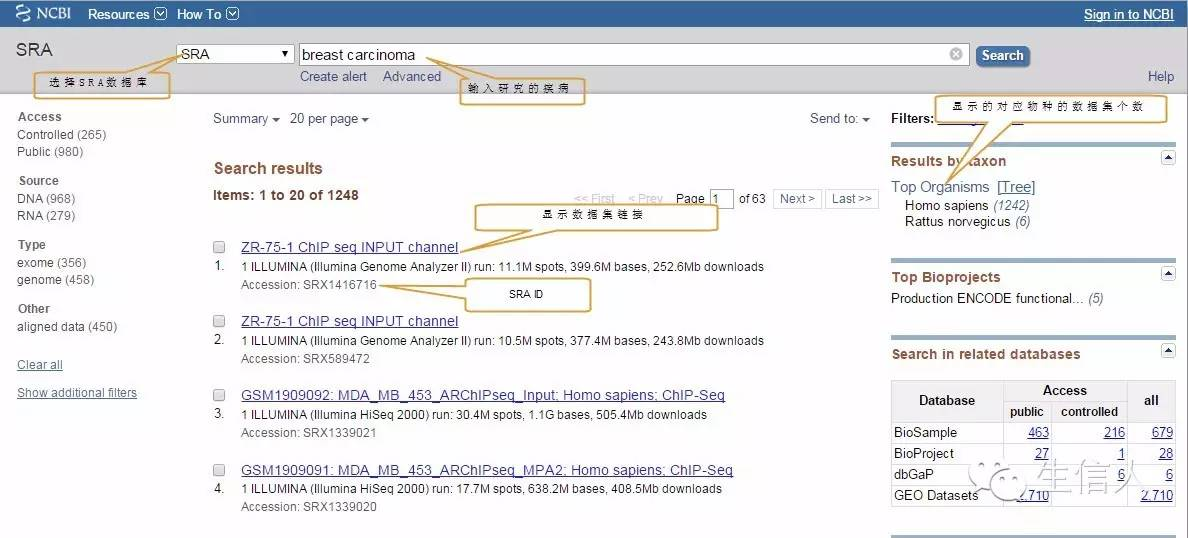
点击第一个案例进入详细信息界面
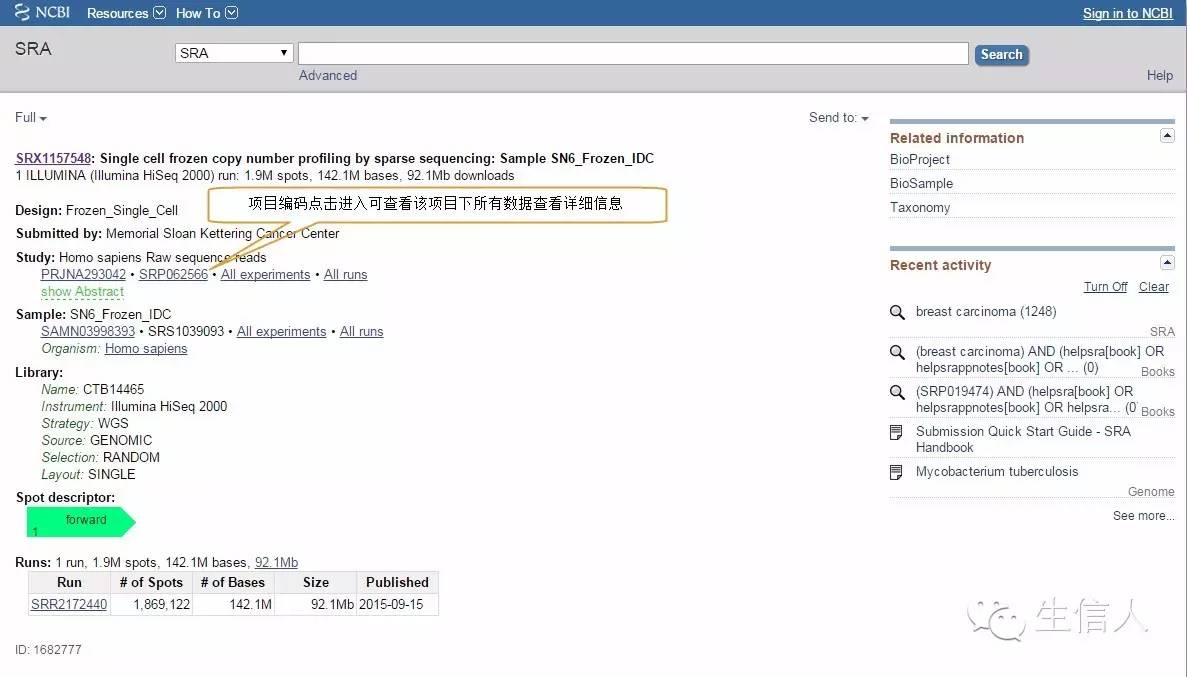
Study详细信息页面
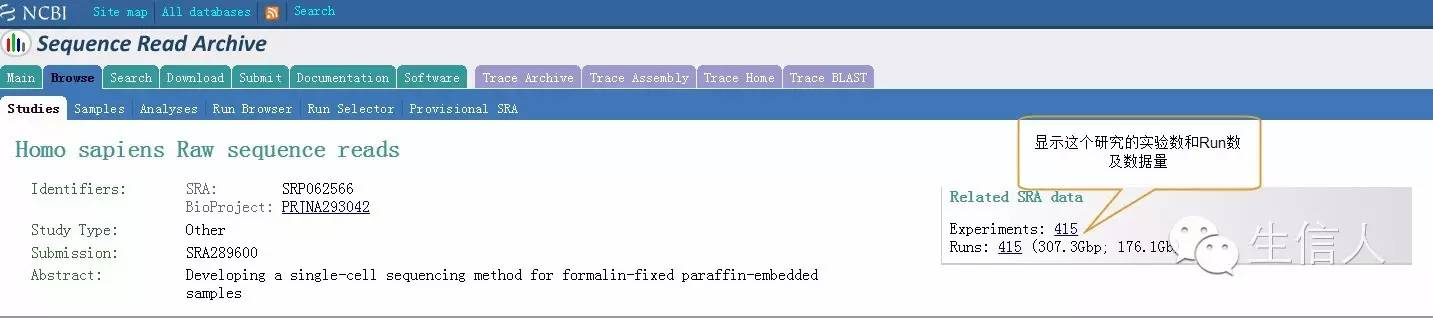
Experiments详细信息页面
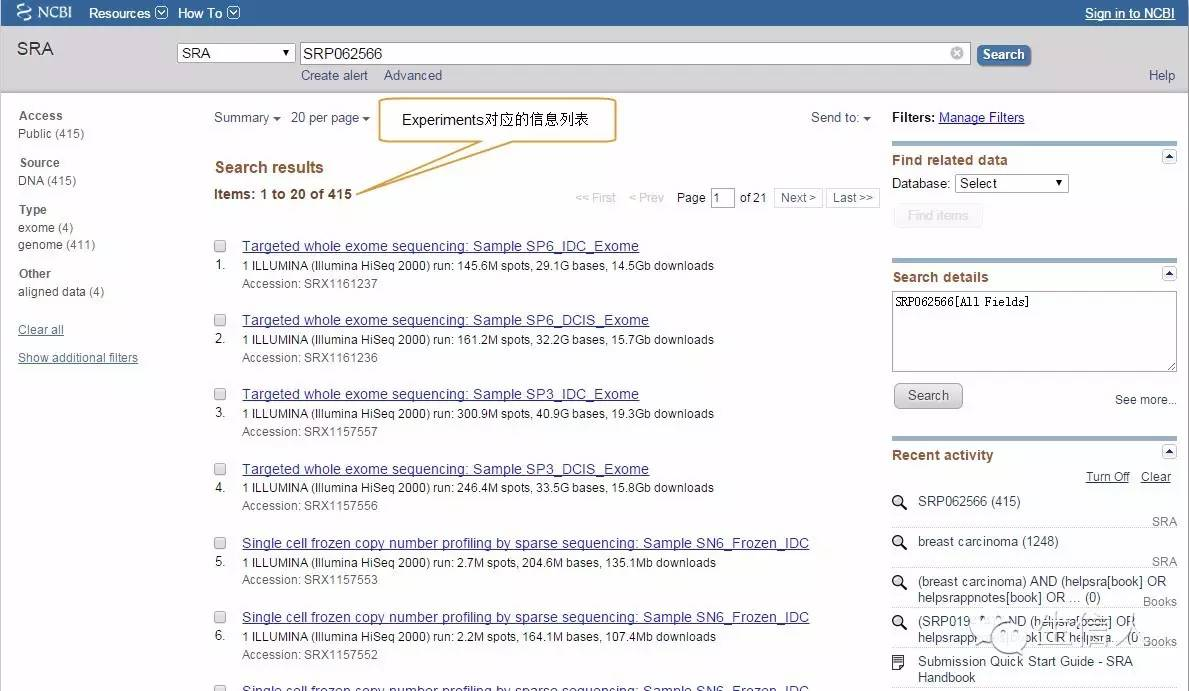
Runs详细信息页面,选择要下载的Runs
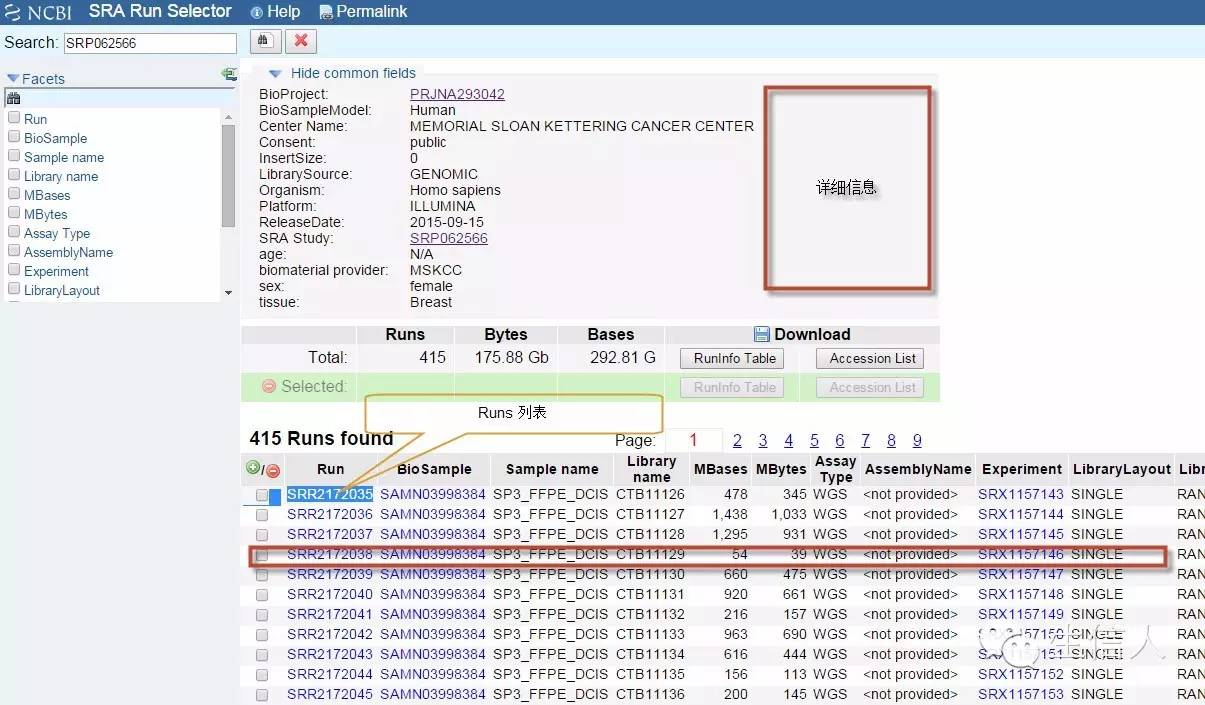
3、下载数据
要下载SRA数据,我们需要先安装SRA Toolkit软件包,下载地址:
https://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software
根据自己的环境下载相应的软件包。
主要包括:
-
CentOS 32/64
-
Ubuntu 32/64
-
MacOS 32/64
-
MS Windows 32/64
以CentOS为例:
1、下载安装:
wget "http://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/current/sratoolkit.current-centos_linux64.tar.gz"
tar xzf sratoolkit.current-centos_linux64.tar.gz
2、运行下载
cd sratoolkit.2.5.7-centos_linux64/bin
./prefetch SRR2172038
下载完成后,会在你的工作主目录下生成一个ncbi的文件夹。
cd ncbi/public/sra
查看SRR2172038.sra数据
3、转换fastq
/sratoolkit.2.5.7-centos_linux64/bin/fastq-dump ./SRR2172038.sra
4、转换fasta
/sratoolkit.2.5.7-centos_linux64/bin/fastq-dump --fasta ./SRR2172038.sra
批量下载SRA数据
1.新建文件,命令为prefetch_bash.sh (感觉命名简单粗暴啊)
vi prefetch_bash.sh
#!/bin/bash
for id in $(seq 1 5) #记住该语法
do
prefetch SRR35899${id}
done
3.给文件一个可执行权限
chmod +755 frefetch_bash.sh
4.添加环境变量或者将其move到/usr/bin即可
添加环境变量:
vi ~/.profile
export PATH=/home/lmt/biosoft/data:$PATH
保存之后需source .profile
5.利用prefetch_bash.sh批量下载所需的SRR文件
在终端输入:prefetch_bash.sh
下载的SRR数据默认存放在:/home/lmt/ncbi/public/sra里