Samtools merge 时出错:
弄了个软链接,如图2,使用链接过去的文件时出现图1的错误,主要是软链接的时候没有全部提供绝对路径。
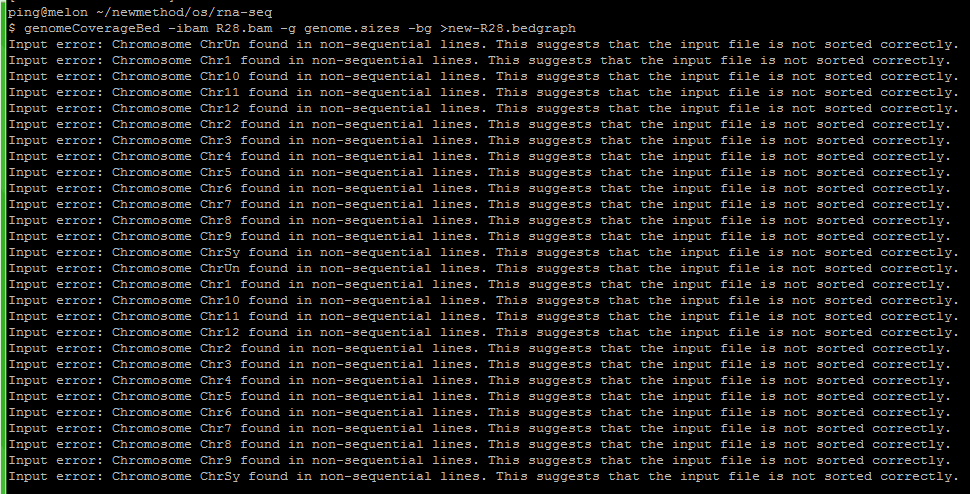
出现以下错误,是因为事先没提前对bam文件进行sort,genomeCoverageBed中的输入文件同样需要经过排序,如若是bed,则须用sort排序,如若是bam,则用samtools sort进行排序。
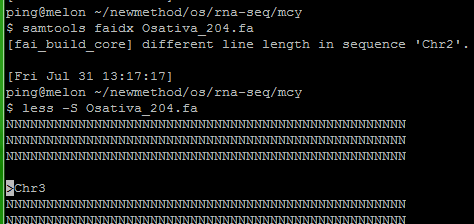
Samtools faidx时出错:
出现以下错误是因为fasta文件中二号染色体和三号染色体间多出了一空行