Haplotype-based GWAS(单倍型全基因组关联分析)是基于 haplotype (单倍型)进行的关联分析,在基因组层面寻找与表型相关的变异。
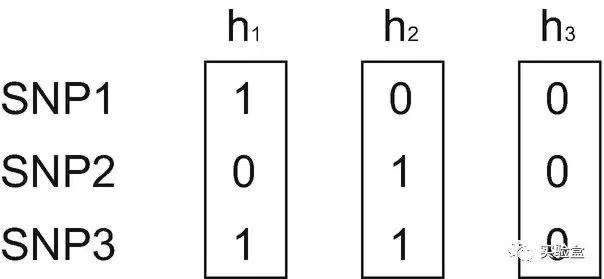
Haplotype 是由一条染色体或线粒体上紧密连锁的多个等位基因组合形成, 每一种组合方式即为一种 haplotype。比如,上图中有 3 个 SNP 和 3 条 染色体,形成了 h1、h2、h3 三种 haplotypes。与单个 SNP 相比,haplotype 提供了更多信息,对定位 causal variant 非常有帮助。
Haplotype-based GWAS 可以用 HAPSTAT、PLINK v1.07、WHAP 等工具进行,不过这些操作通常比较麻烦。另外,haplotypes 数量会随 SNP 数量增大而急剧增大,这会导致检验统计量的自由度非常大,限制了检验的功效。
这里,推荐一个比较新的方法:RAINBOW。它无需 haplotype 的先验信息,把 haplotype 作为 SNP-set (多个 SNPs 组成的集合)处理,分析非常方便。
安装
RAINBOW 这个方法已经集成在 R 包 RAINBOWR 中。RAINBOWR 不仅可以做基于 SNPs 集的GWAS(SNP-set GWAS),也可以做单个 SNP 的 GWAS(Single-SNP GWAS)、分析上位效应(SNP-set x SNP-set interaction)、绘制曼哈顿图和 QQ Plot,功能非常多。
RAINBOWR 稳定版可直接通过 CRAN 安装,最新版则需要通过 GitHub(https://github.com/KosukeHamazaki/RAINBOWR):
#### 稳定版 RAINBOW ####
install.packages("RAINBOWR")
#### 最新版 RAINBOW ####
install.packages("devtools")
devtools::install_github("KosukeHamazaki/RAINBOW")
RAINBOWR 依赖其他的一些包,如果安装过程报错,检查一下这些包是否有安装:Rcpp (win 用户则需 Rtools), rgl, tcltk, Matrix, cluster, MASS, pbmcapply, optimx, methods, ape, stringr, pegas, ggplot2, ggtree, scatterpie, phylobase, haplotypes, ggimage。
RAINBOWR 安装失败通常是因为个别包自动安装失败,需要按提示手动安装缺失的包。其中, ggtree 需从 Bioconductor 安装:BiocManager::install("ggtree")。
数据格式
分析需要三个文件,分别是记录每个个体基因型的文件(geno_score)、基因型位置信息文件(geno_map) 以及表型文件(pheno)。注意,下面的演示例子中,第一行为 header,第一列是行名。
基因型文件
基因型文件 geno_score 需要将每个基因型编码为 -1、0、1 的形式,如果按 additive model 计算的话, -1 代表祖先纯合子,0 代表杂合子,1 代表突变纯合子。每一列为一个个体,每一行为一个 SNP 在不同个体中的基因型:
snp L1 L2 L3 L4 L5 L6
id1000223 1 1 -1 -1 -1 -1
id1000556 -1 -1 1 1 -1 1
id1000673 -1 1 -1 1 -1 1
id1000830 -1 1 1 1 1 1
id1000955 -1 1 1 1 -1 1
id1001073 -1 -1 -1 1 1 -1
如果不知道怎么转换出基因型文件,可用 plink 的 --recode A 编码成 0、1、2 的方式,用 R 读取结果文件后让每个数字减去 1,再转置一下数据框。
基因型位置信息文件
基因型位置信息文件 geno_map 包含每一个 SNP 的名字、染色体和物理位置:
snp marker chr pos
id1000223 id1000223 1 420422
id1000556 id1000556 1 655693
id1000673 id1000673 1 740153
id1000830 id1000830 1 913806
id1000955 id1000955 1 1041748
id1001073 id1001073 1 1172387
表型文件
表型文件 pheno 每一行为一个人,每一列为一个表型,可以包含多个表型:
id Flowering.time.at.Arkansas Flowering.time.at.Faridpur
L1 75.08333333 64
L3 89.5 66
L4 94.5 67
L5 87.5 70
L6 89.08333333 73
L7 105 <NA>
分析
以 RAINBOWR 的实例数据为例:
加载包:
require(RAINBOWR)
读取实例数据:
data("Rice_Zhao_etal")
Rice_geno_score <- Rice_Zhao_etal$genoScore
Rice_geno_map <- Rice_Zhao_etal$genoMap
Rice_pheno <- Rice_Zhao_etal$pheno
过滤 MAF < 0.05 的 SNPs:
x.0 <- t(Rice_geno_score)
MAF.cut.res <- MAF.cut(x.0 = x.0, map.0 = Rice_geno_map)
x <- MAF.cut.res$x
map <- MAF.cut.res$map
选择分析的表型:
trait.name <- "Flowering.time.at.Arkansas"
y <- Rice_pheno[, trait.name, drop = FALSE]
整合转换数据:
modify.data.res <- modify.data(pheno.mat = y, geno.mat = x, map = map,
return.ZETA = TRUE, return.GWAS.format = TRUE)
pheno.GWAS <- modify.data.res$pheno.GWAS
geno.GWAS <- modify.data.res$geno.GWAS
ZETA <- modify.data.res$ZETA
进行单个 SNP 的 GWAS,以前 4 个 主成分和亲缘关系 ZETA 进行校正,并绘制曼哈顿图和 QQ 图:
normal.res <- RGWAS.normal(pheno = pheno.GWAS, geno = geno.GWAS,
plot.qq = TRUE plot.Manhattan = TRUE
ZETA = ZETA, n.PC = 4, P3D = TRUE, count = FALSE)
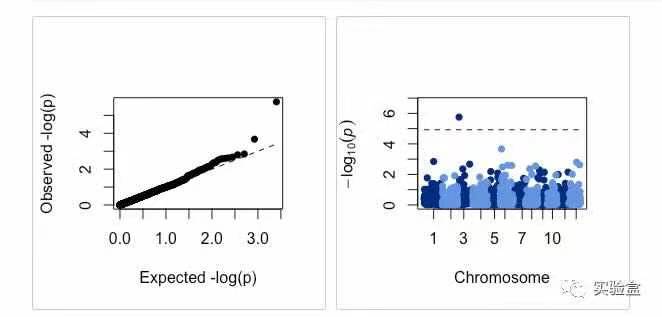
进行 SNP-Set GWAS,以前 4 个 主成分和亲缘关系 ZETA 进行校正,并绘制曼哈顿图和 QQ 图:
SNP_set.res <- RGWAS.multisnp(pheno = pheno.GWAS, geno = geno.GWAS, ZETA = ZETA, plot.qq = TRUE, plot.Manhattan = TRUE, count = FALSE, n.PC = 4, test.method = "LR", kernel.method = "linear", gene.set = NULL, test.effect = "additive", window.size.half = 5, window.slide = 11)
对于一个给定的 SNP,它对应的 SNP set 大小是 2 * window.size.half + 1,也就是说 SNP set 是以一个 SNP 为中心,上下游各自拓展 window.size.half 个SNPs。参数 window.slide 控制了中心 SNP 滑动的步长,如果要 SNPs 分成 bin,可以按 window.slide = 2 * window.size.half + 1 设定。
在上面的这个例子中,SNP-set大小是 11,滑动步长也是 11 个 SNPs。
查看 SNP-Set GWAS结果,结果第 4 列为 负对数转换后的值 -log10(p):
···
See(SNP_set.res$D)
···
SNP-set 也可以通过通过 gene.set 参数自行指定:
gene (or haplotype block) marker
gene_1 id1000556
gene_1 id1000673
gene_2 id1000830
gene_2 id1000955
gene_2 id1001516
… …
FAQ
一
提示 configure: error: gdal-config not found or not executable 报错
需要安装 GDAL(https://gdal.org/index.html),可使用conda安装:conda install -c conda-forge gdal 。
二
安装 ggtree 报错 package ‘ggtree’ is not available for this version of R,是因为 ggtree 需要通过 Bioconductor 安装:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("ggtree")
三
ggimage 安装过程中报错 ERROR: compilation failed for package ‘magick’,需安装 Magick++。可直接用 conda 安装:conda install -c conda-forge imagemagick。
参考
- Multi-SNP haplotype analysis methods for association analysis (DOI: 10.1007/978-1-4939-7274-6_24)
- https://cran.r-project.org/web/packages/RAINBOWR
- https://github.com/KosukeHamazaki/RAINBOWR
