原文链接:http://tecdat.cn/?p=17884
马尔科夫链蒙特卡洛方法
在许多情况下,我们没有足够的计算能力评估空间中所有n维像素的后验概率 。在这些情况下,我们倾向于利用称为Markov-Chain Monte Carlo 算法的程序 。此方法使用参数空间中的随机跳跃来(最终)确定后验分布。MCMC的关键如下:
跳跃概率的比例与后验概率的比例成正比。
跳跃概率可以表征为:
概率(跳跃)*概率(接受)
从长远来看,该链将花费大量时间在参数空间的高概率部分,从而实质上捕获了后验分布。有了足够的跳跃,长期分布将与联合后验概率分布匹配。
MCMC本质上是一种特殊类型的随机数生成器,旨在从难以描述(例如,多元,分层)的概率分布中采样。在许多/大多数情况下,后验分布是很难描述的概率分布。
MCMC使您可以从实际上不可能完全定义的概率分布中进行采样!
令人惊讶的是,MCMC的核心并不难于描述或实施。让我们看一个简单的MCMC算法。
Metropolis-Hastings算法
该算法与模拟退火算法非常相似。
MH算法可以表示为:
Prob(acceptB | A)= min(1,Posterior(B)Posterior(A)⋅Prob(b→a)Prob(a→b))
请注意,从本质上讲,这与“ Metropolis”模拟退火算法相同,后验概率代替了概率,并且 k 参数设置为1。
二元正态例子
请记住,MCMC采样器只是随机数生成器的一种。我们可以使用Metropolis-Hastings采样器来开发自己的随机数生成器,生成进行简单的已知分布。在此示例中,我们使用MH采样器从标准双变量正态概率分布生成随机数。
对于这个简单的示例,我们不需要MCMC采样器。一种实现方法是使用以下代码,该代码从具有相关参数ρ的双变量标准正态分布中绘制并可视化任意数量的独立样本。
-
#################
-
#MCMC采样的简单示例
-
#################
-
-
-
#########
-
# #首先,让我们构建一个从双变量标准正态分布生成随机数的函数
-
-
rbvn<-function (n, rho) #用于从二元标准正态分布中提取任意数量的独立样本。
-
{
-
x <- rnorm(n, 0, 1)
-
y <- rnorm(n, rho * x, sqrt(1 - rho^2))
-
cbind(x, y)
-
}
-
-
#########
-
# 现在,从该分布图中绘制随机抽样
-
bvn<-rbvn(10000,0.98)
-
par(mfrow=c(3,2))
-
plot(bvn,col=1:10000

-
###############
-
# #Metropolis-Hastings双变量正态采样器的实现...
-
-
library(mvtnorm) # 加载一个包,该包使我们能够计算mv正态分布的概率密度
-
-
metropoli<- function (n, rho=0.98){ # 双变量随机数生成器的MCMC采样器实现
-
mat <- matrix(ncol = 2, nrow = n) # 用于存储随机样本的矩阵
-
x <- 0 # 所有参数的初始值
-
-
-
prev <- dmvnorm(c(x,y),mean=c(0,0),sig
-
# 起始位置分布的概率密度
-
mat[1, ] <- c(x, y) # 初始化马尔可夫链
-
-
newx <- rnorm(1,x,0.5) # 进行跳转
-
-
-
newprob <- dmvnorm(c(newx,newy),sigma =
-
# 评估跳转
-
ratio <- newprob/prev # 计算旧位置(跳出)和建议位置(跳到)的概率之比。
-
-
prob.accept <- min(1,ratio) # 决定接受新跳跃的概率!
-
-
-
if(rand<=prob.accept){
-
x=newx;y=newy # 将x和y设置为新位置
-
mat[counter,] <- c(x,y) # 将其存储在存储阵列中
-
-
-
prev <- newprob # 准备下一次迭代
-
然后,我们可以使用MH采样器从该已知分布中获取随机样本…
-
###########
-
# 测试新的M-H采样器
-
-
bvn<-metropoli(10000,0.98)
-
par(mfrow=c(3,2))
-
plot(bvn,col=1:10000)
-
plot(bvn,type=
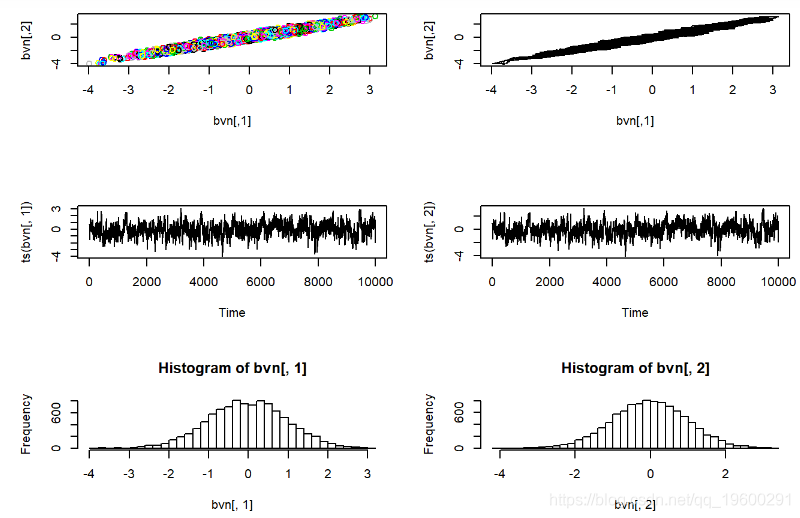
让我们尝试解决一个问题。
MCMC对粘液瘤病进行调查
-
############
-
#黏液病示例的MCMC实现
-
############
-
-
-
-
subset(MyxDat,grade==1
-
-
## grade day titer
-
## 1 1 2 5.207
-
## 2 1 2 5.734
-
## 3 1 2 6.613
-
## 4 1 3 5.997
-
## 5 1 3 6.612
-
## 6 1 3 6.810
选择使用Gamma分布。这是经验分布:
-
###########
-
# 第100次可视化粘液病数据
-
hist(Myx$titer,freq=FALSE)
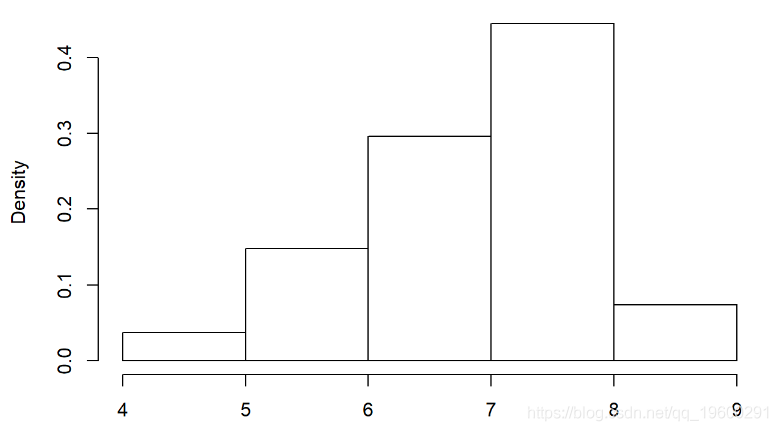
我们需要估算最适合此经验分布的伽马速率和形状参数。这是适合此分布的Gamma的一个示例:
-
#########
-
# ...覆盖生成模型的数据(伽玛分布)
-
-
-
curve(dgamma(x,shape=40,scale=0.15),add=T,col="red")
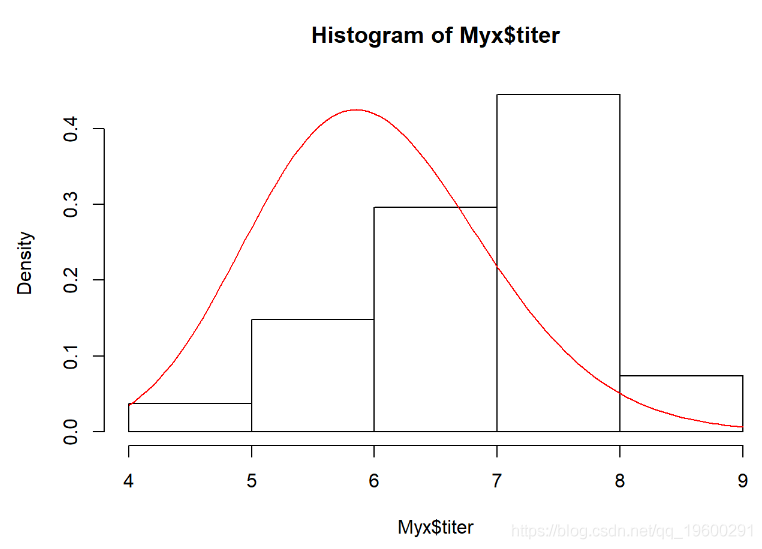
二维(对数)似然面:
-
##############
-
# 定义二维参数空间
-
##############
-
-
shapevec <- seq(3,100,by=0.1)
-
scalevec <- seq(0.01,0.5,by=0.001)
-
-
##############
-
# #定义参数空间内此网格上的似然面
-
##############
-
-
GammaLogLikelihoodFunction <- function(par
-
-
}
-
surface2D <- matrix(nrow=length(shapevec),ncol=length(scalevec)) #初始化存储变量
-
-
newparams <- c(sha
-
-
surface2D[i,j] <- GammaLogLikelihoodFunction(newparams)
-
-
-
-
############
-
# 可视化似然面
-
############
-
-
-
contour(x=shapevec,y=scalevec,z=surface2D,levels=c(-30,-40,-80,-500),add=T)
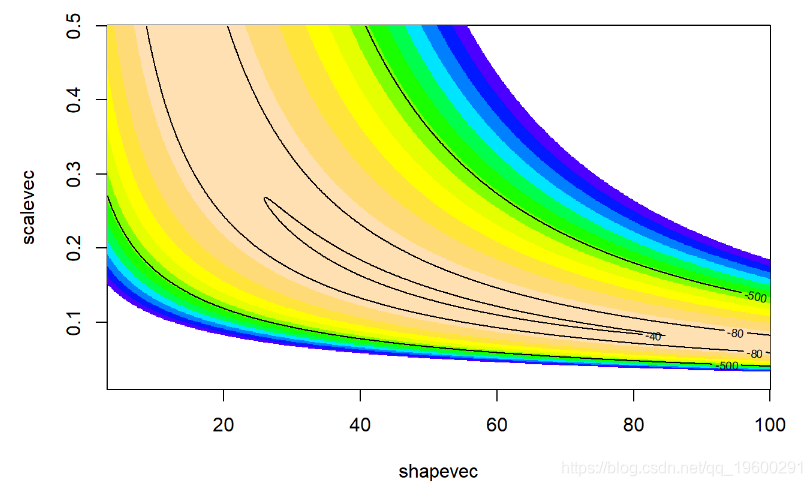
这是MH算法的实现,用于找到后验分布!
首先,我们需要一个似然函数–这次,我们将返回真实概率–而不是对数转换的概率
-
############
-
#编写非对数转换的似然函数
-
-
GammaLike- function(params){
-
prod(dgamma(Myx$titer,shape=params['shape']
-
-
params <- c(shape=40,
-
## shape scale
-
## 40.00 0.15
GammaLike(params)## [1] 2.906766e-22GammaLogLike(params)## [1] -49.58983然后,我们需要预先分配参数!在这种情况下,我们分配gamma(shape = 0.01,scale = 100)和gamma(shape = 0.1,scale = 10)的分布(均值为1且方差略低):
-
#############
-
# 函数返回参数空间中任意点的先验概率密度
-
GammaPriorFunction <- function(params){
-
prior <- c(shape=NA,scale=NA)
-
],3,100)
-
# prior['scale'] <- dunif(params['
-
-
GammaLogPriorFunction <- function(params){
-
prior <- c(shape=NA,scale=NA)
-
'],shape=0.001,scale=1000,log=T)
-
# prior['shape'] <- dunif(params['shape'],3,100)
-
# prior['scale'] <- dunif(params['
-
-
curve(dgamma(x,shape=0.01,scale=1000),3,100)
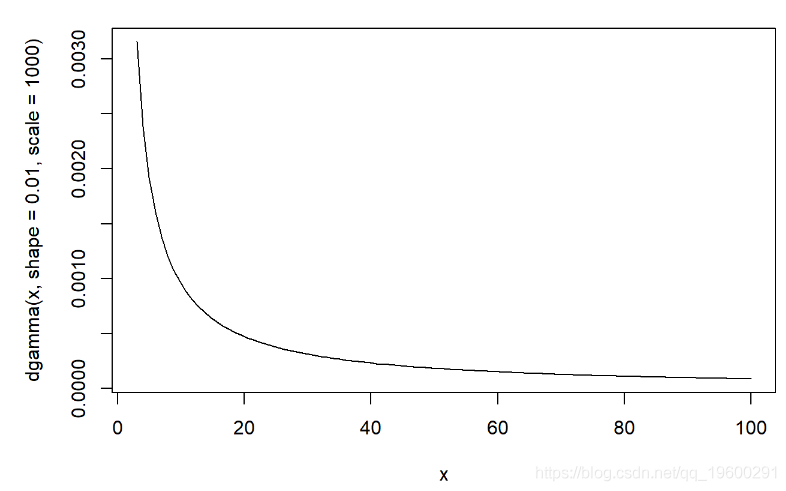
params-
## shape scale
-
## 40.00 0.15
GammaPrior(params)## [1] 1.104038e-06-
prior2D <- matrix(nrow=length(shapevec),ncol=length(scalevec)) # 初始化存储变量
-
-
newparams <- c(shape=50,scale=0
-
for(j in 1:length(scalevec)){
-
newparams['scale'] <- sca
-
-
-
-
############
-
# 可视化似然面
-
############
-
-
image(x=shapevec,y=scalevec,z=prior2D,zlim
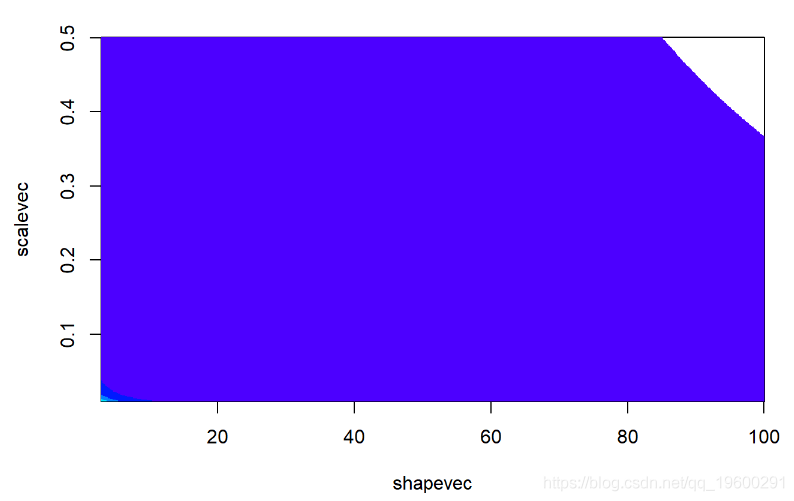
contour(x=shapevec,y=scalevec,z=prior2D,levels=c(-30,-40,-80,-500),add=T)我们假设形状和比例 在先验中是 独立的(联合先验的乘法概率)。然而,并没有对后验参数相关性提出相同的假设,因为概率可以反映在后验分布中。
然后,我们需要一个函数,该函数可以计算参数空间中任何给定跳转的后验概率比率。因为我们正在处理 后验概率的 比率,所以 我们不需要计算归一化常数。
无需归一化常数,我们只需要计算加权似然比(即先验加权的似然比)
-
############
-
# 函数用于计算参数空间中任意两点之间的后验密度比
-
-
PosteriorRatio <- function(oldguess,newguess
-
oldLik <- max(1e-90,GammaLikelihoodFunction(oldguess)) # 计算旧猜测的可能性和先验密度
-
-
newLik <- GammaLikelihoodFunction(newguess) # 在新的猜测值下计算可能性和先验密度
-
-
return((newLik*newPrior)/(oldLik*oldPrior)) # 计算加权似然比
-
}
-
-
PosteriorRatio2 <- function(oldguess,newguess){
-
oldLogLik <- GammaLogLikelihoodFunction(oldguess) # 计算旧猜测的可能性和先验密度
-
-
newLogLik <- GammaLogLikelihoodFunction(newguess) # 在新的猜测值下计算可能性和先验密度
-
-
return(exp((newLogLik+newLogPrior)-(oldLogLik+oldLogPrior))) # 计算加权似然比
-
}
-
-
## [1] 0.01436301PosteriorRatio2(oldguess,newguess)## [1] 0.01436301然后,我们需要一个函数进行新的推测或在参数空间中跳转:
-
############
-
# 为参数空间中的跳转定义:使用正态分布函数进行新的推测
-
newGuess <- function(oldguess)
-
-
jump <- c(shape=rnorm(1,mean=0,sd=sdshapejump),scale=rnorm(1,0,sdscalejump))
-
newguess <- abs(oldguess + ju
-
-
}
-
# 在原始推测附近设置新的推测
-
-
newGuess(oldguess=params)
-
## shape scale
-
## 35.7132110 0.1576337
newGuess(oldguess=params)-
## shape scale
-
## 45.1202345 0.2094243
newGuess(oldguess=params)-
## shape scale
-
## 42.87840436 0.08152061
现在,我们准备实现Metropolis-Hastings MCMC算法:
我们需要一个初始点:
-
##########
-
# 在参数spacer中设置起点
-
-
startingvals <- c(shape=75,scale=0.28) # 算法的起点
测试函数
-
###########
-
# 尝试我们的新函数
-
-
newguess <- newGuess(startingvals) # 在参数空间中跳跃
-
newguess
-
## shape scale
-
## 73.9663949 0.3149796
PosteriorRatio2(startingvals,newguess) # 后验比例差异## [1] 2.922783e-57现在让我们看一下Metropolis-Hastings:
-
###############
-
#可视化Metropolis-Hastings
-
-
chain.length <- 11
-
gth,ncol=2)
-
colnames(guesses) <- names(startingvals)
-
guesses[1,] <- startingvals
-
counter <- 2
-
-
post.rat <- PosteriorRatio2(oldguess,newguess)
-
prob.accept <- min(1,post
-
-
oldguess <- newguess
-
guesses[coun
-
-
-
-
#可视化
-
-
contour(x=shapevec,y=scal
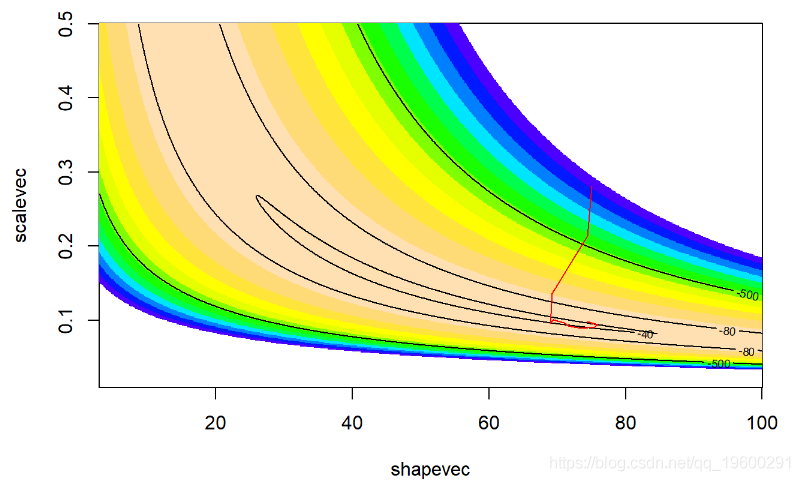
我们运行更长的时间
-
##########
-
# 获取更多MCMC示例
-
-
chain.length <- 100
-
oldgu
-
-
counter <- 2
-
while(counter <= chain.length){
-
newguess <- newGuess(oldguess)
-
post.rat <- Posterio
-
-
rand <- runif(1)
-
if(rand<=prob.accept){
-
ewguess
-
counter=counte
-
-
#可视化
-
-
image(x=shapevec,y=scalevec,z=su
-
urface2D,levels=c(-30,-40,-80,-5
-
lines(guesses,col="red")
更长的时间
-
############
-
#更长的时间
-
-
-
-
chain.length <- 1000
-
oldguess <- startingvals
-
chain.length,ncol=2)
-
colnames(guesses) <- names(startingvals)
-
guesses[1,] <- startingva
-
ess)
-
post.rat <- PosteriorRatio2(oldguess,newguess)
-
prob.accept <- min(1,post.rat)
-
rand <- runif(1)
-
-
guesse
-
-
-
#可视化
-
-
image(x=shapevec,y=scalevec,
-
rface2D,levels=c(-30,-40,-80,-500),a
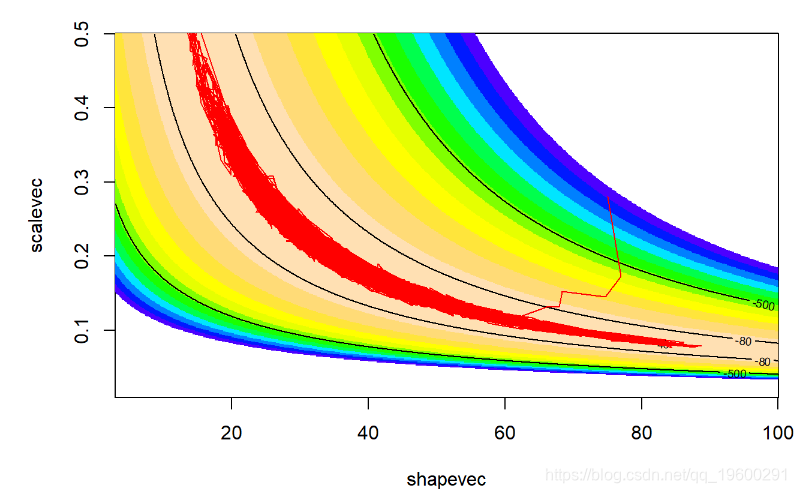
看起来更好!搜索算法可以很好地找到参数空间的高似然部分!
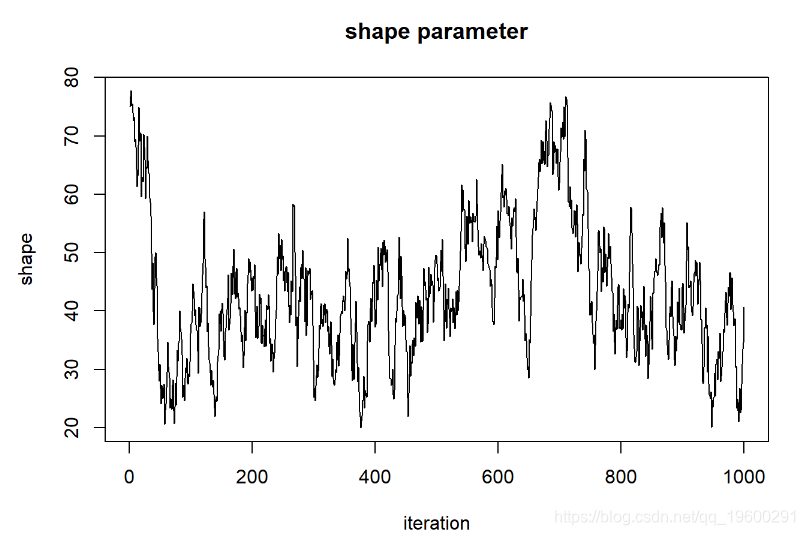
现在,让我们看一下“ shape”参数的链
-
#############
-
# 评估MCMC样本的“轨迹图” ...
-
-
##### Shape 参数
-
-
plot(1:chain.length,guesses[,'sha
对于比例参数
-
###### 比例参数
-
-
plot(1:chain.length,guesses[,'scale'],type="l
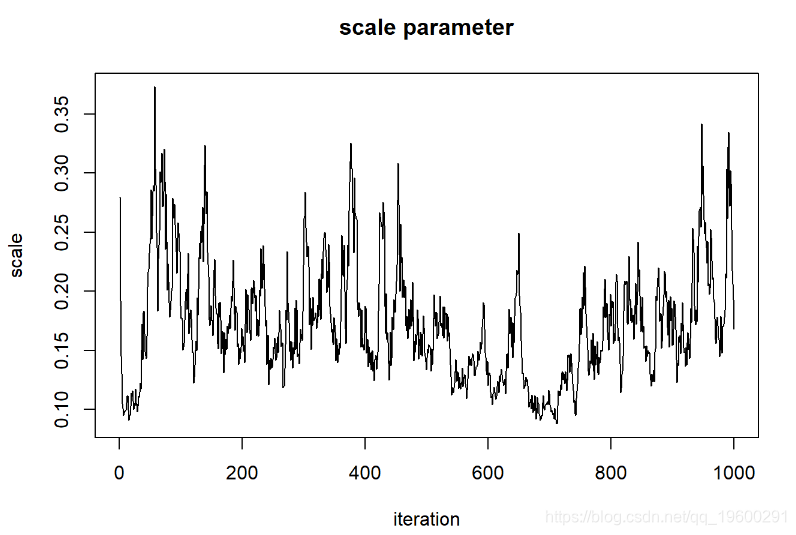
我们可以说这些链已经收敛于形状参数的后验分布吗?
首先,链的起点“记住”起始值,因此不是固定分布。我们需要删除链的第一部分。
-
############
-
# 删除预烧期(允许MCMC有一段时间达到后验概率)
-
-
burn.in <- 100
-
MCMCsamples <- guesses[-c(1:burn.in),]
-
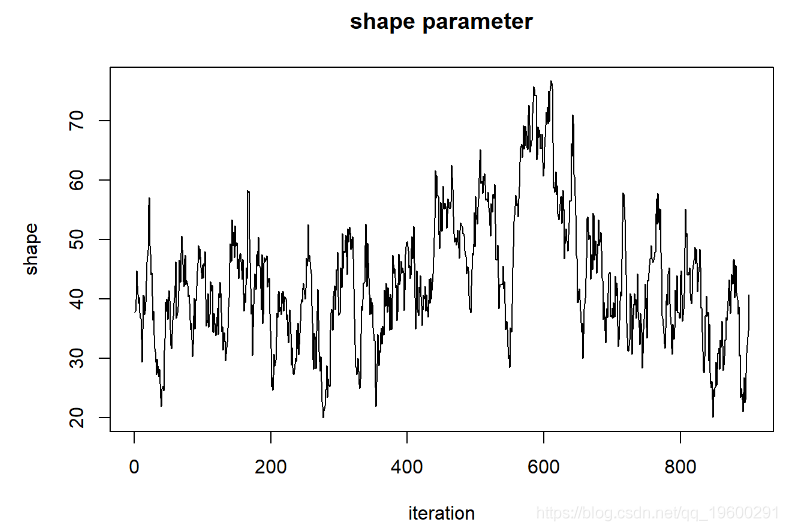
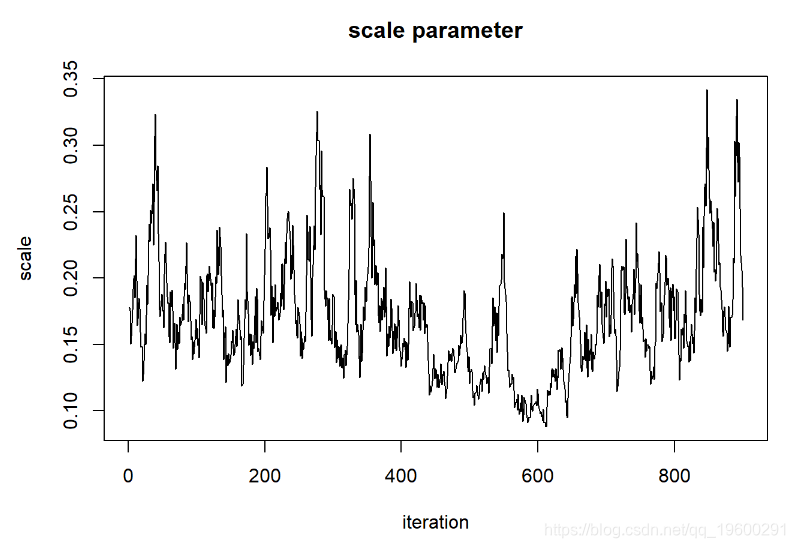
但这看起来还不是很好。让我们运行更长的时间,看看是否得到的东西看起来更像是随机数生成器(白噪声)
-
##########
-
# 再试一次-运行更长的时间
-
-
chain.length <- 20000
-
oldguess <- startingv
-
o2(oldguess,newguess)
-
prob.accept <- mi
-
-
让我们首先删除前5000个样本作为预烧期
-
#############
-
# 使用更长的“预烧”
-
-
burn.in <- 5000
-
MCMCsamples <- guesses[-c(1:bur
现在,让我们再次看一下链条
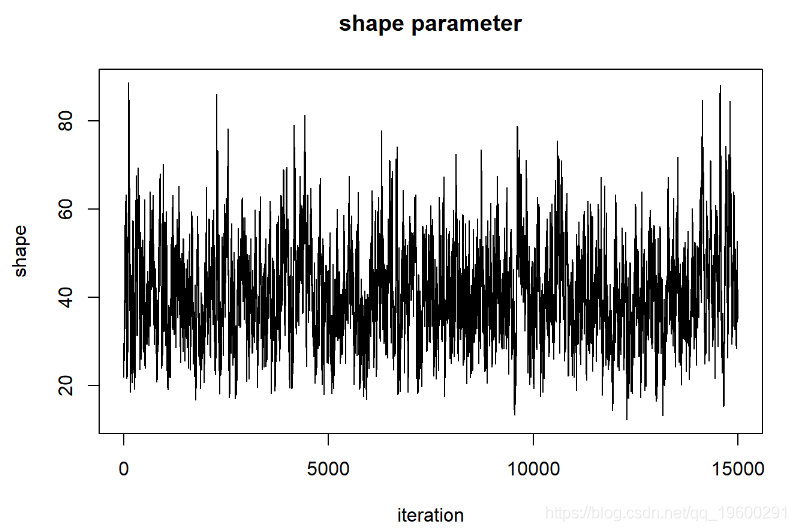
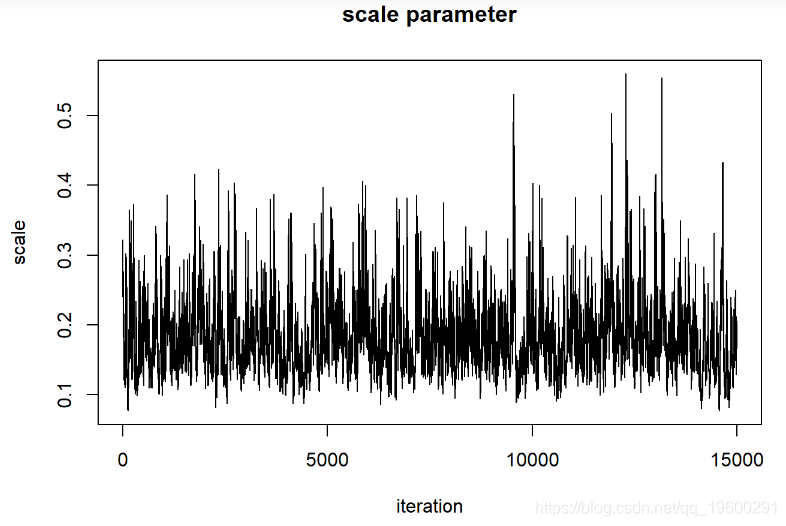
在评估这些迹线图时,我们希望看到看起来像白噪声的“平稳分布”。该轨迹图看起来可能具有一些自相关。解决此问题的一种方法是稀疏MCMC样本:
-
##########
-
# “稀疏” MCMC样本
-
-
thinnedMCMC <- MCMCsamples[seq(1,chain.length,by=5),]
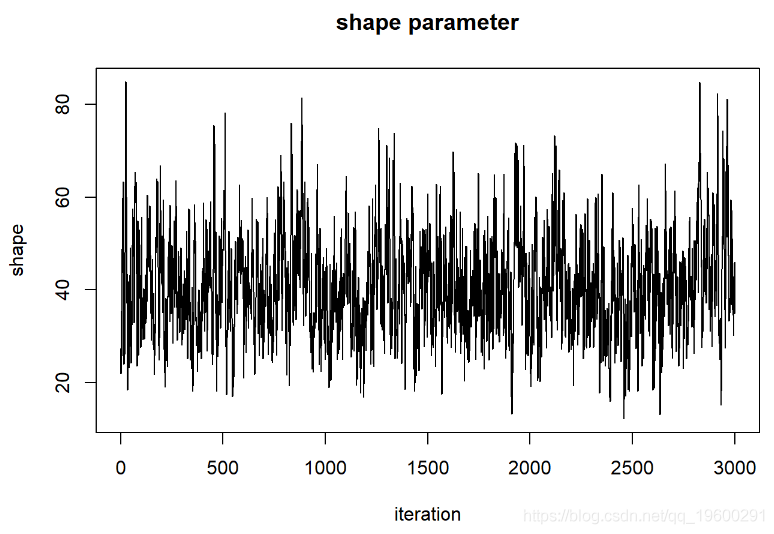
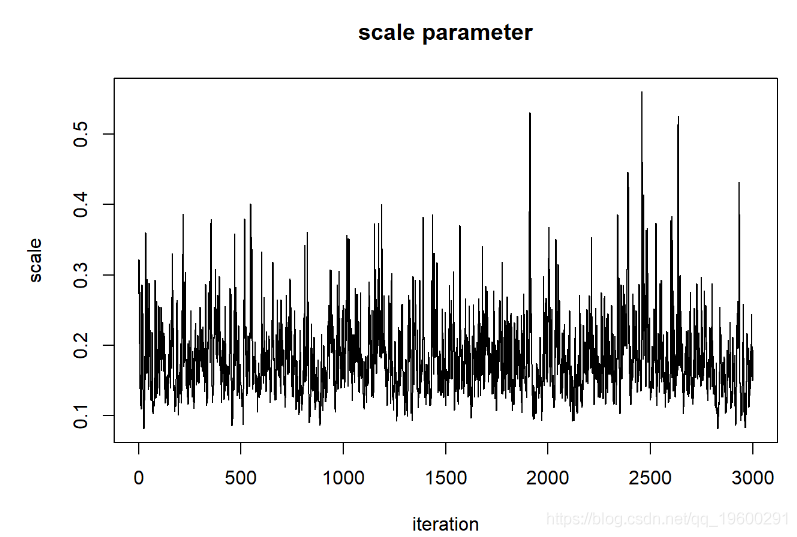
现在我们可以检查我们的后验分布!
-
# 可视化后验分布
-
-
plot(density(thinnedMCMC[,'scale'])
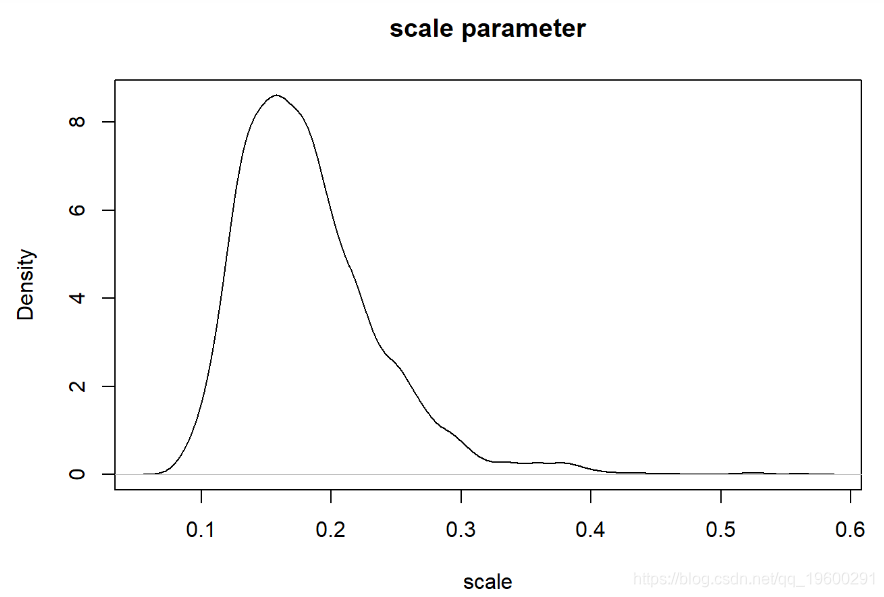
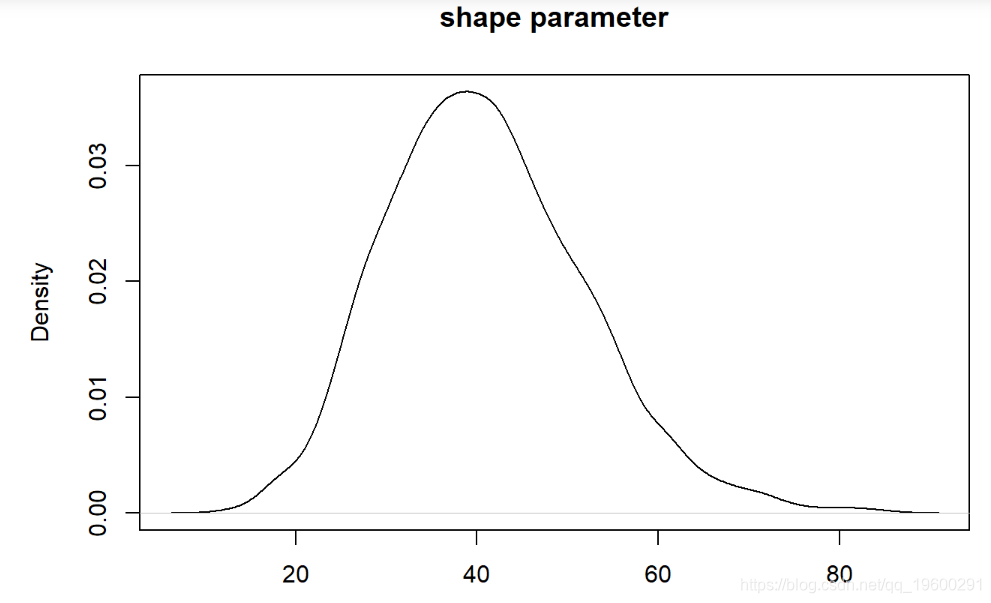
我们可以像以前一样可视化。
-
#########
-
# 更多后验概率检察
-
-
par(mfrow=c(3,2))
-
plot(thinnedMCMC,col=1:10000)
-
plot(thinnedMCMC,type="l")
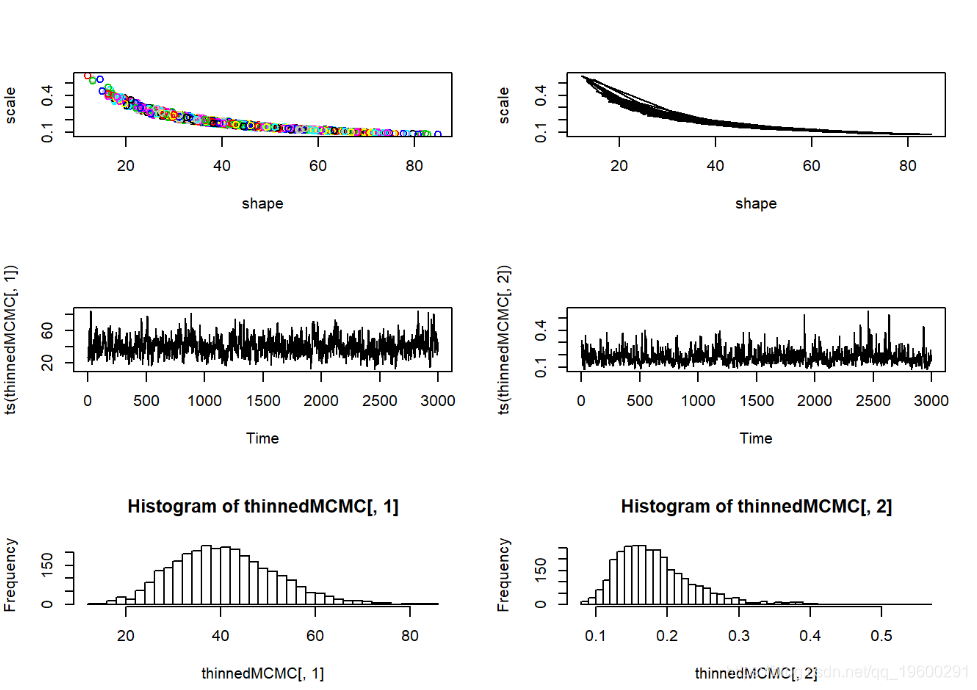
可以修改Metropolis-Hastings MCMC方法来拟合任意模型的任意数量的自由参数。但是,MH算法本身不一定是最有效和灵活的。在实验中,我们使用吉布斯采样,大多采用建模语言 BUGS 。
注意:BUGS实现(例如JAGS)实际上倾向于结合使用MH和Gibbs采样,MH和Gibbs采样器并不是唯一的MCMC例程。例如,“ stan”使用MH采样的一种改进形式,称为“ Hamiltonian Monte Carlo”。
吉布斯Gibbs采样器
Gibbs采样器非常简单有效。基本上,该算法从完整的条件 概率分布(即, 在模型中所有其他参数的已知值作为条件的条件下,对任意参数i的后验分布)中进行 连续采样 。
在很多情况下,我们不能直接制定出我们的模型后验分布,但我们 可以 分析出条件后验分布。尽管如此,即使它在分析上不易处理,我们也可以使用单变量MH程序作为最后方法。
问:为什么Gibbs采样器通常比纯MH采样器效率更高?
二元正态例子
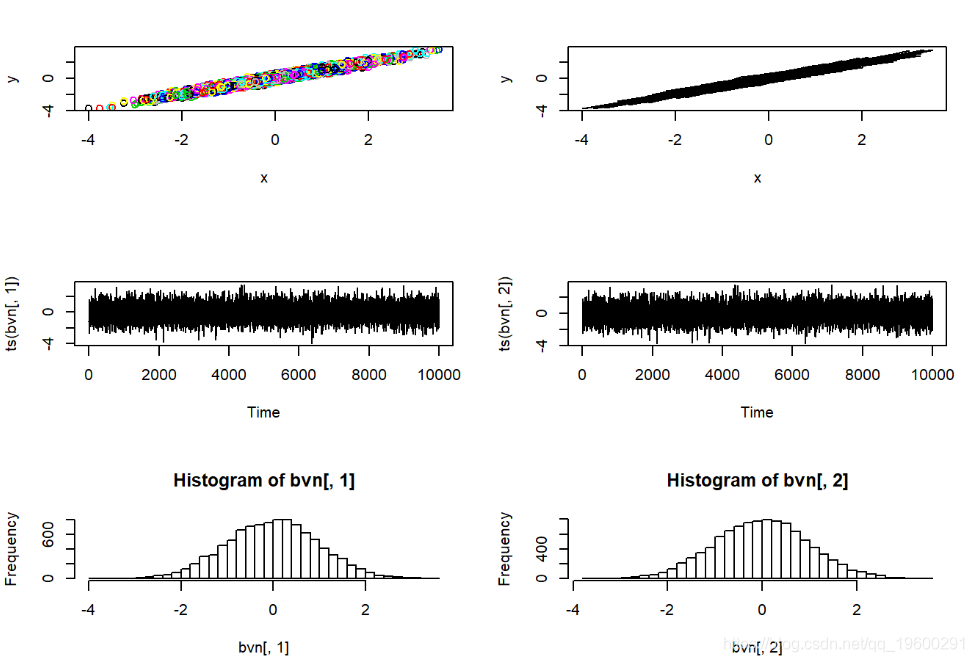
MCMC采样器只是随机数生成器的一种。我们可以使用Gibbs采样器来开发自己的随机数生成器,以实现相当简单的已知分布。在此示例中,我们使用Gibbs采样器从标准双变量正态概率分布生成随机数。注意,吉布斯采样器在许多方面都比MH算法更简单明了。
-
#############
-
#Gibbs采样器的简单示例
-
#############
-
-
########
-
# 首先,回顾一下我们简单的双变量正态采样器
-
rbvn<-function (n, rho){ #f函数用于从双变量标准正态分布中提取任意数量的独立样本。
-
x <- rnorm(n, 0, 1)
-
y <- rnorm(n, rho * x, sqrt(1 - rho^2))
-
-
#############
-
# 现在构造一个吉布斯采样器
-
gibbs<-function (n, rho){ # 双变量随机数生成器的gibbs采样器实现
-
mat <- matrix(ncol = 2, nrow = n) # 用于存储随机样本的矩阵
-
-
mat[1, ] <- c(x, y) # initialize the markov chain
-
for (i in 2:n) {
-
x <- rnorm(1, rho * y, sqrt(1 - rho^2)) # 以y为条件的x中的样本
-
y <- rnorm(1, rho * x, sqrt(1 - rho^2)) # 以x为条件的y中的样本
-
mat[i, ] <- c(x, y)
-
然后,我们可以使用Gibbs采样器从该已知分布中获取随机样本…
-
##########
-
# 测试吉布斯采样器
-
-
-
-
plot(ts(bvn[,2]))
-
hist(bvn[,1],40)
-
hist(bvn[,2],40)
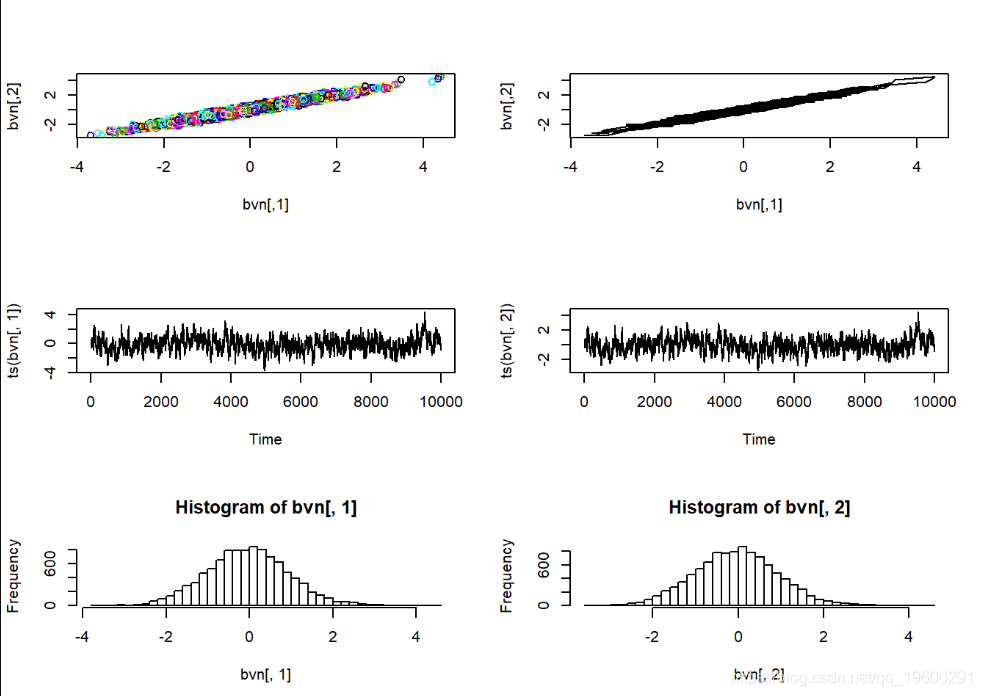
在这里,马尔可夫链的样本中有很多明显的自相关。Gibbs采样器经常有此问题。
示例
BUGS语言
最后,让我们为我们最喜欢的粘瘤病示例创建一个Gibbs采样器,为此,我们将使用BUGS语言(在JAGS中实现)来帮助我们!
JAGS,全称是Just another Gibbs sampler,是基于BUGS语言开发的利用MCMC来进行贝叶斯建模的软件包。它没有提供建模所用的GUI以及MCMC抽样的后处理,这些要在其它的程序软件上来处理,比如说利用R包(rjags)来调用JAGS并后处理MCMC的输出。JAGS相对于WinBUGS/OpenBUGS的主要优点在于平台的独立性,可以应用于各种操作系统,而WinBUGS/OpenBUGS只能应用于windows系统;JAGS也可以在64-bit平台上以64-bit应用来进行编译。
BUGS语言看起来与R类似,但是有几个主要区别:
- 首先,BUGS是一种编译语言,因此代码中的操作顺序并不重要
- BUGS不是矢量化的-您需要使用FOR循环
- 在BUGS中,几个概率分布的参数差异很大。值得注意的是,正态分布通过均值和精度(1 / Variance )进行参数化。
这是用BUGS语言实现的粘液病示例:
-
-
model {
-
-
#############
-
# 似然
-
############
-
for(obs in 1:n.observations){
-
titer[obs] ~ dgamma(shape,rate
-
-
#############
-
# 先验
-
############
-
-
-
-
rate <- 1/scale # 将BUGS的scale参数转换为“ rate”
-
}
我们可以使用R中的“ cat”函数将此模型写到您的工作目录中的文本文件中:
-
###########
-
# BUGS建模语言中的粘液瘤示例
-
-
##########
-
# 将BUGS模型写入文件
-
-
cat("
-
model {
-
-
#############
-
# 似然
-
############
-
for(obs in 1:n.observations){
-
titer[obs] ~ dgamma(shape,rate)
-
-
-
-
#############
-
# 先验
-
############
-
shape ~ dgamma(0.001,0.001
-
-
file="BUGSmodel.txt")
现在我们已经将BUGS模型打包为文本文件,我们将数据捆绑到一个列表对象中,该列表对象包含BUGS代码中引用的所有相关数据:
-
############
-
# 将数据封装到单个“列表”对象中
-
-
myx.data <- list(
-
-
n.observations = length(Myx$titer
-
## $titer
-
## [1] 5.207 5.734 6.613 5.997 6.612 6.810 5.930 6.501 7.182 7.292 7.819
-
## [12] 7.489 6.918 6.808 6.235 6.916 4.196 7.682 8.189 7.707 7.597 7.112
-
## [23] 7.354 7.158 7.466 7.927 8.499
-
##
-
## $n.observations
-
## [1] 27
然后,我们需要为所有参数定义初始值。将其定义为一个函数很方便,因此可以使用不同的起始值来初始化每个MCMC链。
-
###########
-
# 用于为所有自由参数生成随机初始值的函数
-
-
-
-
-
init.vals.for.bugs()
-
## $shape
-
## [1] 51.80414
-
##
-
## $scale
-
## [1] 0.2202549
init.vals.for.bugs()-
## $shape
-
## [1] 89.85618
-
##
-
## $scale
-
## [1] 0.2678733
init.vals.for.bugs()-
## $shape
-
## [1] 69.22457
-
##
-
## $scale
-
## [1] 0.1728908
现在我们可以调用JAGS!
-
###########
-
# 运行 JAGS
-
##########
## Loading required package: rjags-
## The following object is masked from 'package:coda':
-
##
-
## traceplot
-
-
-
jags.fit <- run.jags(model="BUGSmodel.txt",
-
-
## Compiling rjags model...
-
## Calling the simulation using the rjags method...
-
## Adapting the model for 100 iterations...
-
## Running the model for 5000 iterations...
-
## Simulation complete
-
## Finished running the simulation
-
jags.fit) # 转换为“ MCMC”对象(CODA包)
-
-
-
##
-
## Iterations = 101:5100
-
## Thinning interval = 1
-
## Number of chains = 3
-
## Sample size per chain = 5000
-
##
-
## 1. Empirical mean and standard deviation for each variable,
-
## plus standard error of the mean:
-
##
-
## Mean SD Naive SE Time-series SE
-
## shape 51.6013 14.36711 0.1173070 2.216657
-
## scale 0.1454 0.04405 0.0003597 0.006722
-
##
-
## 2. Quantiles for each variable:
-
##
-
## 2.5% 25% 50% 75% 97.5%
-
## shape 28.76333 40.9574 50.1722 60.2463 82.7532
-
## scale 0.08346 0.1148 0.1378 0.1687 0.2389
plot(jagsfit.mcmc)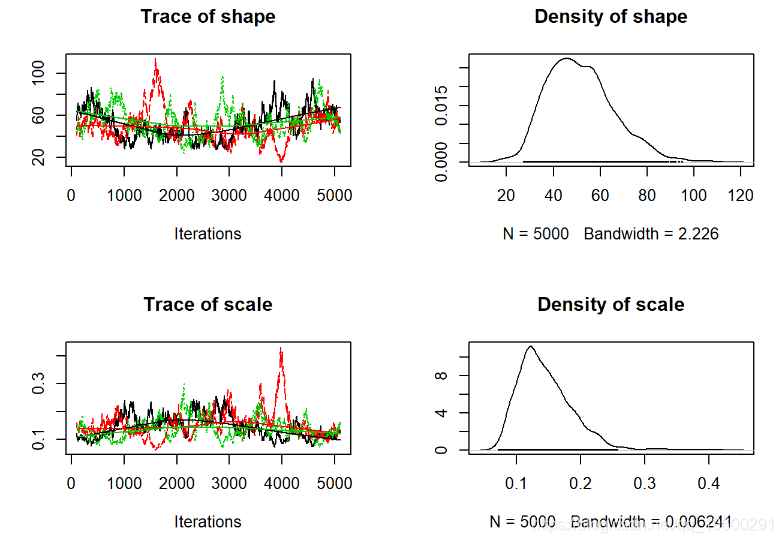
评估收敛
第一步是视觉检查-我们寻找以下内容来评估收敛性:
- 当视为“轨迹图”时,每个参数的链应看起来像白噪声(模糊的毛毛虫)或类似的噪声
- 多个具有不同起始条件的链条看起来应该相同
我们可能在这里可以做得更好的一种方法是使链条运行更长的时间,并丢弃初始样本我们还可以。
通过细化链来尝试减少自相关,我们每20个样本中仅保留1个。
-
################
-
#运行链更长时间
-
-
jags.fit <-
-
sample = 10000,n.chains = 3,adapt = 1000,burnin = 1000,
-
summarise = FALSE,thin=20,method = "parallel" )
-
## Calling 3 simulations using the parallel method...
-
## Following the progress of chain 1 (the program will wait for all
-
## chains to finish before continuing):
-
## Welcome to JAGS 4.2.0 on Wed Oct 23 11:33:35 2019
-
## JAGS is free software and comes with ABSOLUTELY NO WARRANTY
-
## Loading module: basemod: ok
-
## Loading module: bugs: ok
-
## . . Reading data file data.txt
-
## . Compiling model graph
-
## Resolving undeclared variables
-
## Allocating nodes
-
## Graph information:
-
## Observed stochastic nodes: 27
-
## Unobserved stochastic nodes: 2
-
## Total graph size: 37
-
## . Reading parameter file inits1.txt
-
## . Initializing model
-
## . Adapting 1000
-
## -------------------------------------------------| 1000
-
## ++++++++++++++++++++++++++++++++++++++++++++++++++ 100%
-
## Adaptation successful
-
## . Updating 1000
-
## -------------------------------------------------| 1000
-
## ************************************************** 100%
-
## . . . Updating 200000
-
## -------------------------------------------------| 200000
-
## ************************************************** 100%
-
## . . . . Updating 0
-
## . Deleting model
-
## .
-
## All chains have finished
-
## Simulation complete. Reading coda files...
-
## Coda files loaded successfully
-
## Finished running the simulation
-
jagsfit.mcmc <- as.mcmc.list
-
# 转换为“ MCMC”对象(CODA包)
-
-
##
-
## Iterations = 2001:201981
-
## Thinning interval = 20
-
## Number of chains = 3
-
## Sample size per chain = 10000
-
##
-
## 1. Empirical mean and standard deviation for each variable,
-
## plus standard error of the mean:
-
##
-
## Mean SD Naive SE Time-series SE
-
## shape 47.1460 12.9801 0.0749404 0.292218
-
## scale 0.1591 0.0482 0.0002783 0.001075
-
##
-
## 2. Quantiles for each variable:
-
##
-
## 2.5% 25% 50% 75% 97.5%
-
## shape 25.14757 37.9988 45.9142 55.1436 75.5850
-
## scale 0.09147 0.1256 0.1509 0.1825 0.2773
plot(jagsfit.mcmc)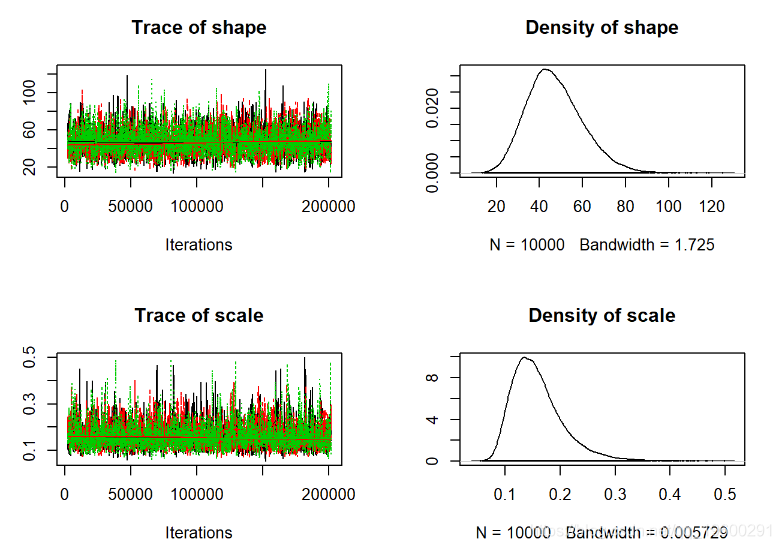
从视觉上看,这看起来更好。现在我们可以使用更多的定量收敛指标。
Gelman-Rubin诊断
一种简单而直观的收敛诊断程序是 Gelman-Rubin诊断程序,该诊断程序基于简单的蒙特卡洛误差来评估链之间是否比应有的链距更大:
-
##############
-
# 运行收敛诊断
-
-
gelman(jagsfit.mcmc)
-
## Potential scale reduction factors:
-
##
-
## Point est. Upper C.I.
-
## shape 1 1.00
-
## scale 1 1.01
-
##
-
## Multivariate psrf
-
##
-
## 1
通常,1.1或更高的值被认为收敛不佳。为模型中的所有可用参数计算GR诊断。如果测试失败,则应尝试运行更长的链!
所以这个模型看起来不错!

最受欢迎的见解
4.R语言中的block Gibbs吉布斯采样贝叶斯多元线性回归