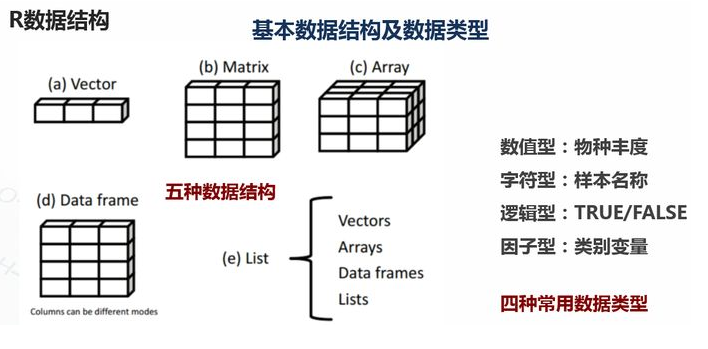

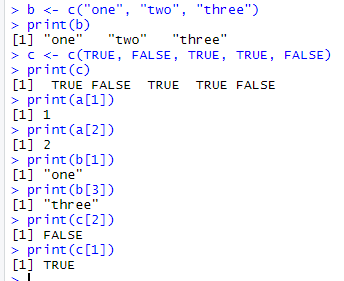
1.基础数据结构 1.1 向量 # 创建向量a a <- c(1,2,3) print(a)


1.2 矩阵 #创建矩阵 mymat <- matrix(c(1:10), nrow=2, ncol=5, byrow=TRUE) #取第二行 mymat[2,] #取第二列 mymat[,2] #第一行第五列的元素 mymat[1,5]


1.3 数组 #创建数组 myarr <- array(c(1:12),dim=c(2,3,2)) print(myarr) #取矩阵或数组的维度 dim(myarr) #取第一个矩阵的第一行第二列 myarr[1,2,1]


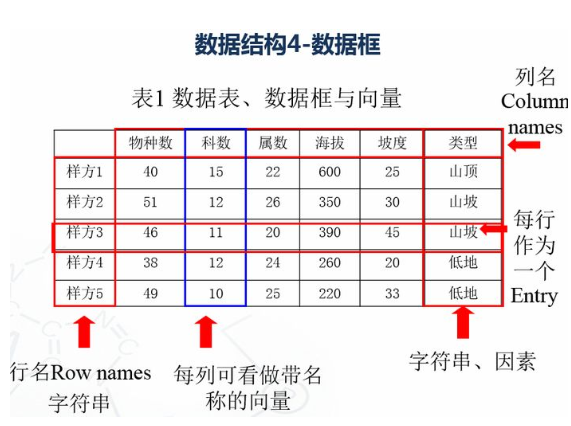
1.4 数据框 # 创建数据框 kids <- c("Wang", "Li") age <- c("18", "16") df <- data.frame(kids, age) print(df) #第一行 df[1,] #第二列 df[,2] #前两行,前两列 df[1:2,1:2] #根据列名称 df$kids #行名称 rownames(df) #列名称 colnames(df) str(df)

1.4.1 因子变量
变量:类别变量,数值变量
类别数据对于分组数据研究非常有用。(男女,高中低)
R中的因子变量类似于类别数据。

1.5 列表 列表以一种简单的方式组织和调用不相干的信息,R函数的许多运行结果都是以列表的形式返回 #创建列表 lis <- list(name='fred',wife='mary',no.children=3,child.ages=c(4,7,9)) print(lis) #列表组件名 lis$name #列表位置访问 lis[[1]]


p <- 0.1 if(p<=0.05){ print("p<=0.05!") }else{ print("p>0.05!") }


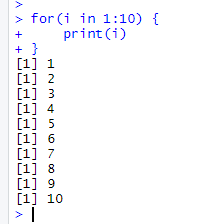
for(i in 1:10) { print(i) }
i <- 1 while(i<10) { print(i) i <- i + 1 }

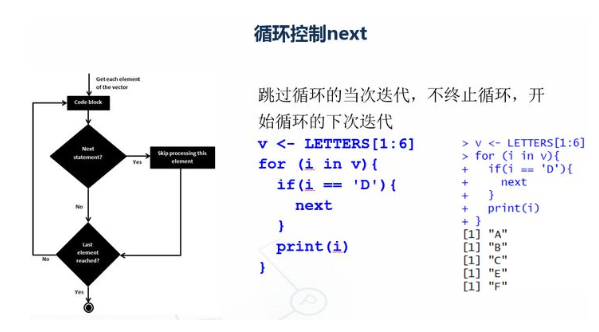
v <- LETTERS[1:6] print(v) for (i in v){ if(i == 'D'){ next } print(i) }

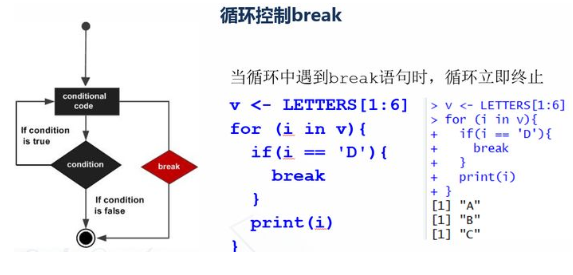
v <- LETTERS[1:6] for (i in v){ if(i == 'D'){ break } print(i) }

2.5 R函数 函数是组织好的,可重复使用的,用来实现单一,或相关联功能的代码段 rcal <- function(x,y){ z <- x^2 + y^2; result <- sqrt(z) ; result; } # 调用函数 rcal(3,4)

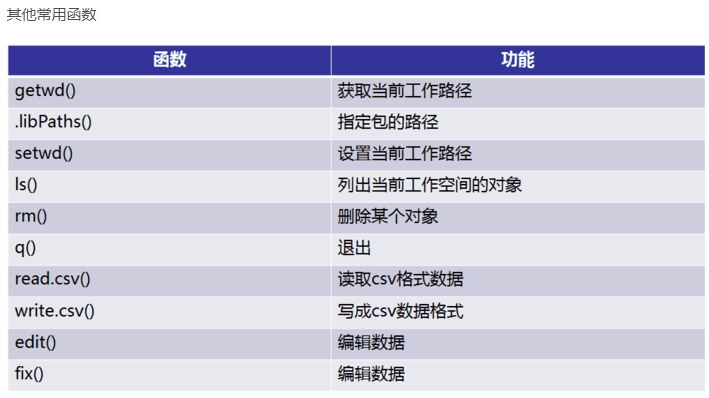
3. 读写数据 #数据读入 getwd() setwd('C:/Users/Administrator/Desktop/file') dir() top <- read.table("otu_table.p10.relative.tran.xls",header=T,row.names=1,sep=' ',stringsAsFactors = F) top10 <- t(top) #数据写出logtop10<-log(top10+0.000001) head(top10, n=2) write.csv(logtop10,file="logtop10.csv", quote=FALSE, row.names = TRUE) write.table(logtop10,file="logtop10.xls",sep=" ", quote=FALSE, row.names = TRUE, col.names = TRUE)

4.1 tidyr包
tidyr包的四个函数
宽数据转为长数据:gather()
长数据转为宽数据:spread()
多列合并为一列: unite()
将一列分离为多列:separate()
library(tidyr) gene_exp <- read.table('geneExp.csv',header = T,sep=',',stringsAsFactors = F) head(gene_exp) #gather 宽数据转为长数据 gene_exp_tidy <- gather(data = gene_exp, key = "sample_name", value = "expression", -GeneID) head(gene_exp_tidy) #spread 长数据转为宽数据 gene_exp_tidy2<-spread(data = gene_exp_tidy, key = "sample_name", value = "expression") head(gene_exp_tidy2)

4.2 dplyr包
dplyr包五个函数用法:
筛选: filter
排列: arrange()
选择: select()
变形: mutate()
汇总: summarise()
分组: group_by()
library(tidyr) library(dplyr) gene_exp <- read.table("geneExp.csv",header=T,sep=",",stringsAsFactors = F) gene_exp_tidy <- gather(data = gene_exp, key = "sample_name", value = "expression", -GeneID) #arrange 数据排列 gene_exp_GeneID <- arrange(gene_exp_tidy, GeneID) #降序加 deschead(gene_exp_GeneID ) #filter 数据按条件筛选 gene_exp_fiter <- filter(gene_exp_GeneID ,expression>10) head(gene_exp_fiter) #select 选择对应的列 gene_exp_select <- select(gene_exp_fiter ,sample_name,expression) head(gene_exp_select)


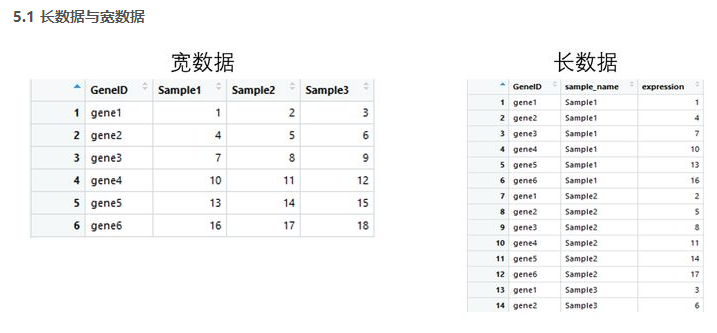
library(tidyr) library(ggplot2) #基础绘图 #宽数据file file <- read.table("geneExp.csv",header=T,sep=",",stringsAsFactors = F,row.names = 1) barplot(as.matrix(file),names.arg = colnames(file), beside =T ,col=terrain.colors(6)) legend("topleft",legend = rownames(file),fill = terrain.colors(6)) #ggplot2绘图 gene_exp <- read.table("geneExp.csv",header=T,sep=",",stringsAsFactors = F) gene_exp_tidy <- gather(data = gene_exp, key = "sample_name", value = "expression", -GeneID) #长数据head(gene_exp_tidy) ggplot(gene_exp_tidy,aes(x=sample_name,y=expression,fill=GeneID)) + geom_bar(stat='identity',position='dodge')
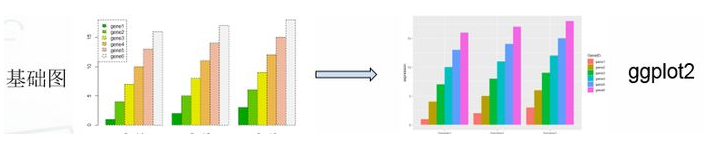

#Rnorm正态分布 个数 平均值 标准差 x <- rnorm(20, 2, 1) y <- rnorm(20, 4, 2) # plot是泛型函数,根据输入类型的不同而变化 #Type p 代表点 l 代表线 b 代表两者叠加 plot(x, y, cex=c(1:3), type="p", pch=19, col = "blue", cex.axis=1.5, col.axis="darkgreen", font.axis=2, main="这是主标题:plot初试", font.main=2, cex.main=2, col.main="green", sub="这是副标题:图1", font.sub=3, cex.sub=1.5, col.sub="red", xlab="这是x轴标签", ylab="这是y轴标签", cex.lab=1.5, font.lab=2, col.lab="grey20", xlim=c(0,3), ylim=c(0,7)) abline(h=2, v=3, lty=1:2, lwd=2,col="red") legend("topright", legend="我是图例 我在这儿",text.col="red", text.width=0.5)

图形参数: 符号和线条:pch、cex、lty、lwd 颜色:col、col.axis、col.lab、col.main、col.sub、fg、bg 文本属性:cex、cex.axis、cex.lab、cex.main、cex.sub、font、font.axis、font.lab、font.main、font.sub 文本添加、坐标轴的自定义和图例 title()、main、sub、xlab、ylab、text() axis()、abline() legend() 多图绘制时候,可使用par()设置默认的图形参数 par(lwd=2, cex=1.5) 图形参数设置: par(optionname=value,…) par(pin=c(width,height)) 图形尺寸 par(mfrow=c(nr,nc)) 图形组合,一页多图 layout(mat) 图形组合,一页多图 par(mar=c(bottom,left,top,right)) 边界尺寸 par(fig=c(x1,x2,y1,y2),new=TURE) 多图叠加或排布成一幅图
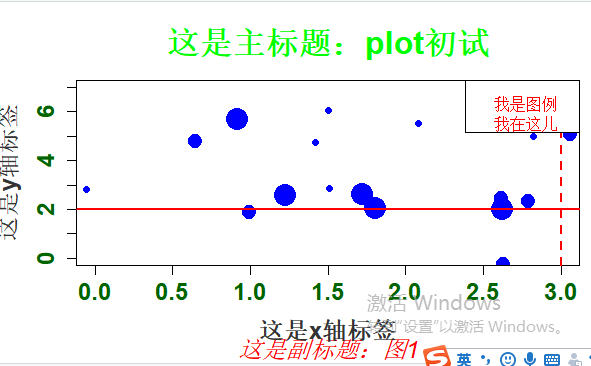
#图形组合: attach(mtcars) #复制当前图形参数设置 opar <- par(no.readonly=TRUE) #设置图形参数 par(mfrow=c(2,2)) layout(matrix(c(1,2,2,3),2,2,byrow=TRUE)) plot(wt,mpg,main="Scatterplot of wt vs mpg") hist(wt,main="Histogram of wt") boxplot(wt,main="Boxplot of wt") #返回原始图形参数detach(mtcars) par(opar)

5.3 柱形图
file <- read.table("barData.csv",header=T,row.names=1,sep=",",stringsAsFactors = F) #转化为矩阵 dataxx <- as.matrix(file) #抽取颜色 cols <- terrain.colors(3) #误差线函数 plot.error <- function(x, y, sd, len = 1, col = "black") { len <- len * 0.05 arrows(x0 = x, y0 = y, x1 = x, y1 = y - sd, col = col, angle = 90, length = len) arrows(x0 = x, y0 = y, x1 = x, y1 = y + sd, col = col, angle = 90, length = len) } x <- barplot(dataxx, offset = 0, ylim=c(0, max(dataxx) * 1.1),axis.lty = 1, names.arg = colnames(dataxx), col = cols, beside = TRUE) box() legend("topright", legend = rownames(dataxx), fill = cols, box.col = "transparent") title(main = "An example of barplot", xlab = "Sample", ylab = "Value") sd <- dataxx * 0.1 for (i in 1:3) { plot.error(x[i, ], dataxx[i, ], sd = sd[i, ]) }
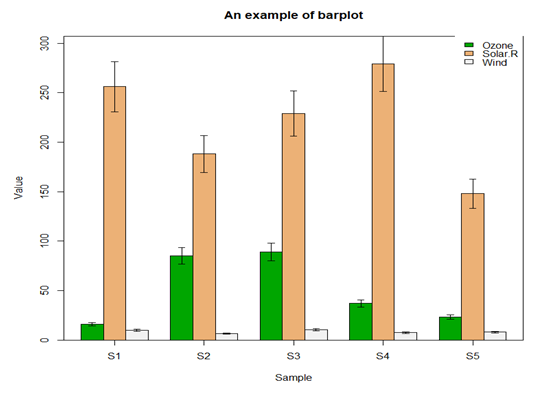
5.4 二元图
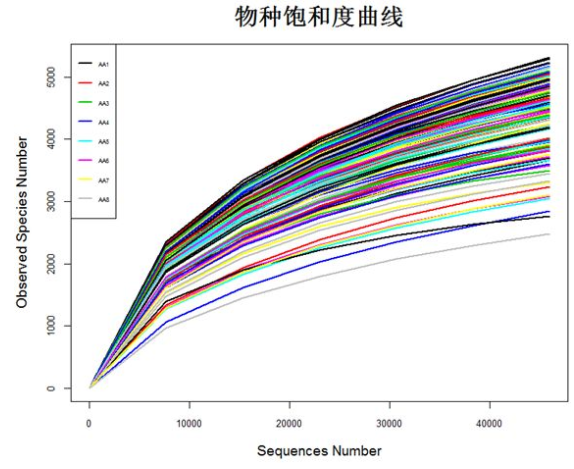
matdata <- read.table("plot_observed_species.xls", header=T) #查看数据属性和结构 tbl_df(matdata) y<-matdata[,2:145] attach(matdata) matplot(series,y, ylab="Observed Species Number",xlab="Sequences Number", lty=1,lwd=2,type="l",col=1:145,cex.lab=1.2,cex.axis=0.8) legend("topleft",lty=1, lwd=2, legend=names(y)[1:8], cex=0.5,col=1:145) detach(matdata)
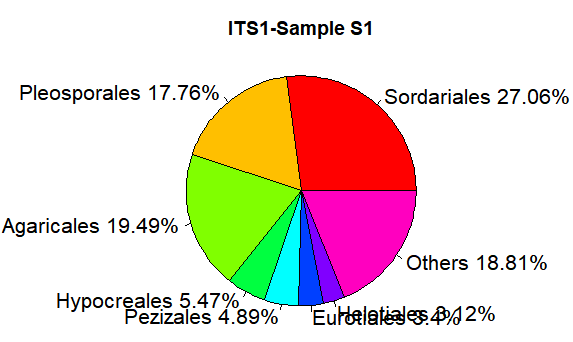
5.5 饼状图
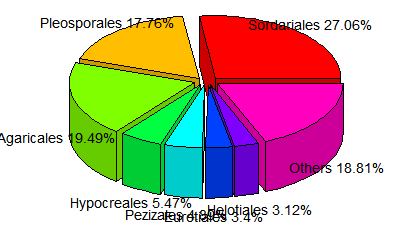
relative <- c(0.270617,0.177584,0.194911,0.054685,0.048903,0.033961, 0.031195,0.188143) taxon <- c("Sordariales","Pleosporales","Agaricales","Hypocreales","Pezizales","Eurotiales","Helotiales","Others") ratio <- round(relative*100,2) ratio <- paste(ratio,"%",sep="") label <- paste(taxon,ratio,sep=" ")
pie(relative,labels=label, main="ITS1-Sample S1",radius=1,col=rainbow(length(label)),cex=1.3)
library(plotrix) fan.plot(relative,labels=label,main="Fan plot")
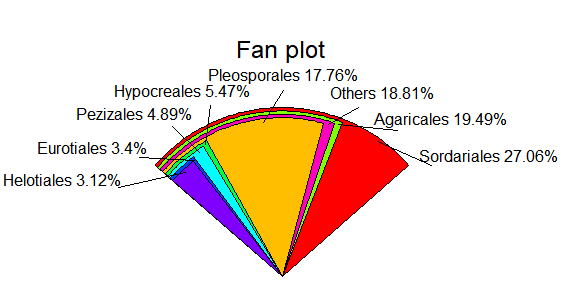
pie3D(relative,labels=label, height=0.2, theta=pi/4, explode=0.1, col=rainbow(length(label)),border="black",font=2,radius=1,labelcex=0.9)
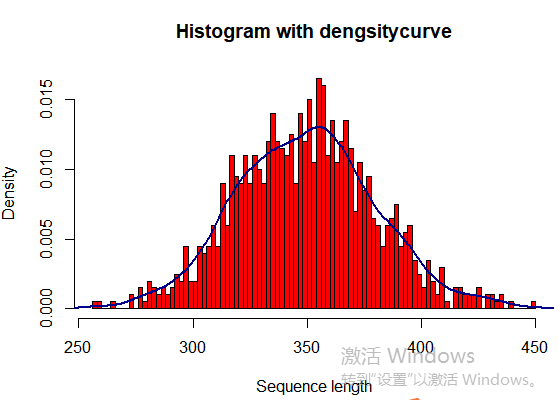
5.6 直方图
seqlength <- rnorm(1000, 350, 30) hist(seqlength,breaks=100,col="red", freq=FALSE, main="Histogram with dengsitycurve", ylab="Density", xlab="Sequence length") lines(density(seqlength),col="blue4",lwd=2)
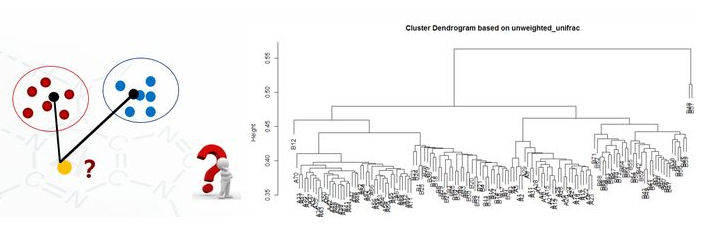
5.7 聚类图
clu <- read.table("unweighted_unifrac_dm.txt", header=T, row.names=1, sep=" ") head(clu) dis <- as.dist(clu) h <- hclust(dis, method="average") plot(h, hang = 0.1, axes = T, frame.plot = F, main="Cluster Dendrogram based on unweighted_unifrac", sub="UPGMA")

#保存图片代码
pdf(file="file.pdf", width=7, height=10) png(file="file.png",width=480,height=480) jpeg(file="file.png",width=480,height=480) tiff(file="file.png",width=480,height=480) dev.off()