二代测序大大提高了测序的通量,伴随而来的基因组也遍地开花,但在构建高质量的参考基因组方面仍然不能令人满意。随着新的测序技术如三代/光学/Hic的出现,使得构建高质量的参考基因组更加高效。对于一些重要的研究对象,一个参考基因组显然是不足以满足研究需求的。以两篇16年发表的人的基因组文献为例,对构建高质量的基因组以及深入分析提供一个案例。
Chinese genome: HX1
HX1是由暨南大学主导完成的中国人参考基因组”华夏一号”,采用三代+光学的测序技术,完成的一个高质量的基因组。
1.测序基本信息
测序材料 中国健康成年男性新鲜抽取的血样,核型正常
测序方法 Pacbio RSII测序仪,P6/C4酶
BioNano Irys测序仪,NT.BspQI酶
HiSeq X测序仪,PE150测序
Iso_Seq测序,构建了四个文库:1-2kb,2-3kb,3-5kb,5kb+
测序数据 RSII测得377个SMRT cells,共得到309Gb测序数据,平均长度7.0kb
Iyrs测得12个cells,平均长度259kb,测序深度101X
HiSeq X测得428.8Gb数据,测序深度143X
Iso_Seq测得50个cells,共得到5.8Gb测序数据
2.基因组概述
首先,通过falcon组装软件对pacbio测序数据进行纠错和拼接,得到5843条contigs,N50长度8.3Mb,基因组大小2.9 Gb;然后,通过illumina测序数据对基因组进行校正;最终,通过光学数据进行混拼,得到scaffold版本基因组,N50长度达到22.0Mb,gap大小39.3Mb,基因组大小2.93Gb。
与GRCh38相比,HX1中包含12.8Mb新的序列,且其中4.1Mb序列未在以前发布的亚洲人群基因组中发现。基于组装的HX1填补GRCh38上的gaps,28.4%(274个)gaps能够被完全填补或部分缩短,共7.1Mb的序列。
通过Iso_Seq数据,共预测到30006个基因座上的58383个同源异构体,其中包括57个未在GENCODE转录组数据库中存在的同源异构体。使用PCR以及Sanger测序,进一步验证了一个预测的新转座元件和两个新基因的准确性。
3.变异分析
在结构变异(SV)检测方面,比较pacbio/illumina/bionano数据分析结果,pacbio长reads检测的敏感性最好,并且某些结构变异包含具有种族特异性的功能元件。其中,通过比较包含在千人基因组计划中的SVs,验证了一个东亚人群特有的缺失变异。
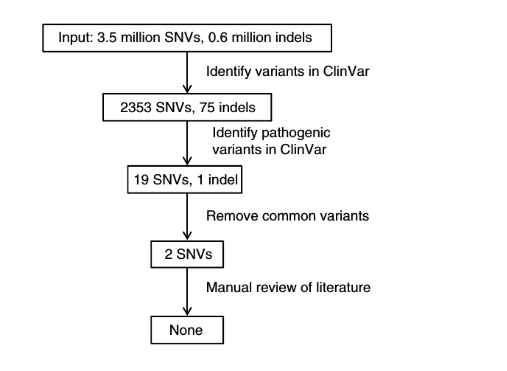
通过illumina测序数据,识别出3518309个单核苷酸变异(SNVs)和625690个插入缺失变异(indels),进一步分析变异的功能和临床相关性。通过过滤最小基因频率(MSF)大于0.01以及注释到dbSNP数据库中的突变,得到新突变信息,其中有372个SNVs和50个indels位于外显子区域;通过将变异注释到ClinVar数据库,得到”致病“分类的变异,进一步通过MSF过滤以及人工查阅文献,证明2个”致病变异“为错误的临床数据,HX1中未发现致病突变。
Korean genome: AK1
AK1是首尔大学完成的韩国人参考基因组“大韩一号”,采用三代+光学的测序数据进行全基因组组装,BAC+10X进行单倍型区分,从而完成的一个高质量的二倍体基因组。对AK1该如何翻译也是思考了一番,是”高丽一号“还是”朝鲜一号“呢?考察了友邦的历史之后,最终选择了高大上的”大韩一号“。
1.测序基本信息
测序材料 AK1个体的永生化细胞系
测序方法 Pacbio RSII测序仪,P6/C4酶
BioNano Irys测序仪,NT.BspQI酶
GemCode构建10X Genomics文库,Hiseq2500测序
HiSeq X测序仪,PE150测序
BAC文库来自62758个BAC clones,PE100测序
Iso_Seq测序,构建了四个文库:1-2kb,2-3kb,3-6kb,5kb+
测序数据 RSII测得380个SMRT cells,测序深度101X
Iyrs测得97X和108X两轮数据,后者用于防止酶切脆性位点的片段化
HiSeq X测序深度72X
Haplotype BACs reads测序深度47X
10X linked reads深度30X
2.基因组概述
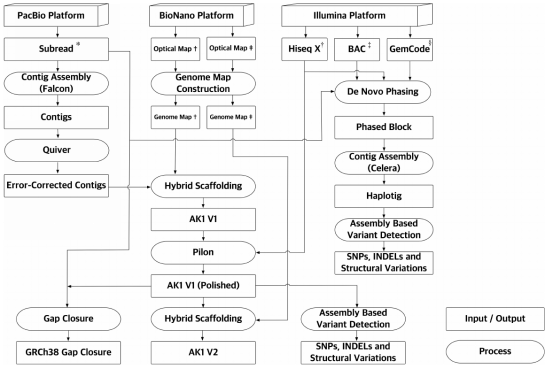
通过falcon组装软件对pacbio测序数据进行拼接,得到3128条contigs,N50长度17.9Mb,基因组大小2.87Gb;经Quiver校正后的基因组结合光学数据进行混拼,得到scaffold版本基因组,scaffold数目2832条,N50长度达到44.8Mb,其中最长的91条scaffold覆盖了基因组的90%,且8条染色体臂被单条scaffold完整跨过,gap大小37.3Mb,基因组大小2.90Gb。
为进一步更好的反映二倍体基因组结构,组装出成对的同源染色体的单倍型结果,首先整合了BACs reads/10X linked reads/pacbio long reads/illumina short reads数据进行单倍型区分,结果显示单倍型的区块N50长度达到11.5Mb;然后,通过分别组装分到不同单倍型的pacbio reads,得到18964条haplotigs,总大小4.8Gb。其中,通过单倍型A和同源区域reads组装的结果为haplotig A,总大小2.63Gb;通过单倍型B的reads组装的结果为haplotig B,总大小2.19Gb。
3.变异分析
在结构变异(SV)检测方面,通过直接比较组装结果,总共发现AK1和GRCh37间存在18210个SVs。其中,发生在外显子区域的615个变异中,427个是新变异,且68%并不影响蛋白功能;对发生变异的31个基因进行功能注释,它们与离子结合、表皮生长因子、纤连蛋白有关。
单倍体的特异突变可以通过比较haplotigs间的变异来识别,这些单倍体间的杂合SNVs/indels/SVs总碱基大小69.8Mb。进一步进行单倍型的验证,通过对人白细胞抗原(HLA)基因的pacbio捕获测序,结果表明基于组装的分型方法能有效的解决高变区的区分。
两篇人的基因组都聚焦于整合不同平台测序数据与高质量参考基因组的构建,对于个人基因组的变异分析也进行了比较全面的概述。
看着有关人类基因组的科学研究在短短二十年内取得的巨大成果,也是感触良多。如果说人类基因组计划标志着人类探索自身奥秘的重要一步,千人基因组计划开启大规模人群基因组遗传多态性的探索,或许个人基因组的时代在下一个十年就会到来。打开基因之书,我们将看到是健康与疾病,还是筛选与偏见?对于疾病相关的检查产品固然是值得期待,但可验性值得商榷,规范化、标准化之路漫漫修远;而消费级的产品的往往涉及个人的隐私,而且大众关于基因的偏见和盲目是显而易见的,“转基因”的事情就可见一斑。
这是最好的时代,也是最坏的时代,这是智慧的时代,也是愚蠢的时代。
PS: 感觉我还是见识太少,“精准美容”之后,"精准辟谷"粉墨登场,令人嗟叹!