Illumina Synthetic Long Read Sequencing Allows Recovery of Missing Sequences even in the “Finished” C. elegans Genome
Illumina合成长读测序允许恢复缺失的序列,甚至在“完成”的秀丽隐杆线虫基因组
Scientific Reports volume 5, Article number: 10814 (2015)
Abstract
Most next-generation sequencing platforms permit acquisition of high-throughput DNA sequences, but the relatively short read length limits their use in genome assembly or finishing. Illumina has recently released a technology called Synthetic Long-Read Sequencing that can produce reads of unusual length, i.e., predominately around 10 Kb. However, a systematic assessment of their use in genome finishing and assembly is still lacking. We evaluate the promise and deficiency of the long reads in these aspects using isogenic C. elegans genome with no gap. First, the reads are highly accurate and capable of recovering most types of repetitive sequences. However, the presence of tandem repetitive sequences prevents pre-assembly of long reads in the relevant genomic region. Second, the reads are able to reliably detect missing but not extra sequences in the C. elegans genome. Third, the reads of smaller size are more capable of recovering repetitive sequences than those of bigger size. Fourth, at least 40 Kbp missing genomic sequences are recovered in the C. elegans genome using the long reads. Finally, an N50 contig size of at least 86 Kbp can be achieved with 24×reads but with substantial mis-assembly errors, highlighting a need for novel assembly algorithm for the long reads.
大多数下一代测序平台可以获得高通量的DNA序列,但相对较短的读取长度限制了它们在基因组组装或整理中的应用。
Illumina公司最近发布了一项被称为合成长读测序的技术,该技术可以产生不寻常长度的阅读,例如,主要是10kb左右。
然而,对于它们在基因组整理和组装中的应用,仍缺乏系统的评估。
我们使用无间隙的等基因秀丽隐杆线虫基因组来评估长序列在这些方面的前景和不足。
首先,read的准确率很高,能够恢复大部分类型的重复序列。
然而,串联重复序列的存在阻止了在相关基因组区域预装配长序列。
第二,读取能够可靠地检测出线虫基因组中缺失而非额外的序列。
第三,较小的读取比较大的读取更能恢复重复序列。
第四,在线虫基因组中,利用长reads至少可以找回40 Kbp缺失的基因组序列。
最后,通过24次读取可以得到至少86 Kbp的N50 contig大小,但存在大量的误组装错误,这就突出了对长时间读取的新型组装算法的需求。
Introduction
The high-throughput sequencing technology, also referred to as Next Generation Sequencing (NGS), has transformed biomedical research from genetics to developmental biology. Capacity of generating large volume of sequencing reads in a short period of time enables genome assembly, genotyping, expression profiling and systematic identification of DNA binding sites in a way that is difficult or impractical otherwise. One critical issue associated with all NGS platforms is the read length on top of the read throughput and accuracy. The relatively short length of sequencing reads produced by most NGS platforms1 limits their use particularly in genome assembly and finishing. For example, ambiguity often remains when the short reads are mapped against a reference genome or among one another, which is further complicated by the accuracy of read sequence. The short length also makes it problematic in variation call and de novo genome assembly. Despite the substantial efforts that have been made in the past decade to increase the read length, for example, from 22 bp to up to 300 bp by Illumina platform2, these lengths are still unsatisfactory for many applications, including de novo genome assembly, genome gap finishing and identification of complex structural variations in a draft genome. Therefore, a tradeoff has to be made across different NGS platforms to balance the read length and yield. For example, the 454 platform can produce reads of length up to 1 Kbp, which is useful in resolving genomic gaps3. Such tradeoff has also catalyzed the third generation sequencing, a term coined for a sequencing method capable of producing reads of unusual length. One example is the single molecule real time sequencing (SMRT) from PacBio, which is able to generate sequencing reads up to 30 Kbp and has been demonstrated to be useful in resolving the complex genomic regions4. However, most of the single-pass reads suffer from a high error rate up to 15-18%5, thus need to be corrected before being used for genome assembly and other applications. The high false positive rate of indels (insertion and deletion) also hinders the use of the PacBio reads in variation calling5.
Illumina has recently released Synthetic Long-Read technology (http://www.illumina.com/products/truseq-synthetic-long-read-kit.ilmn), which allows construction of synthetic long reads from the short sequencing reads generated with its existing HiSeq platform. A surprising long read length plus its high accuracy is posed to affect de novo genome assembly or gap finishing in a draft genome. This technology, also known as Moleculo, has been demonstrated its use by performing de novo genome assembly of the Botryllus schlosseri, a star ascidian animal6 and whole-genome haplotyping of humans7. The long reads have also been used to resolve a subset of highly-repetitive transposons in the genome of Drosophila melanogaster8. However, a systematic evaluation of the reads on their ability in resolving repetitive sequences and identification of complex genomic variations remains to be seen. In particular, the deficiency of the reads in genome finishing and de novo genome assembly has not been characterized. To this end, a high quality of “finished” genome that contains all resolved repetitive sequences will be needed. C. elegans genome is the choice for this task due to its following characteristics. First, its genome is a “finished” one with no gap9, providing an opportunity for unbiased evaluation of the read accuracy and genomic coverage. Second, all types of repetitive sequences have been unambiguously resolved, allowing systematic assessment of the long reads in recovering various repeats. Third, a well annotated C. elegans genome alone with enormous amount of NGS sequencing data permits validation of the potential errors either in the current genome assembly or in the long reads. Finally, the isogenic C. elegans genome suffers little heterozygosity, which is often the issue associated with de novo assembly and variation calling in the genomes of outcrossing organisms. By mapping the long reads and its assembled contigs back to the C. elegans genome, we systemically characterized the promise and deficiency of the reads in genome improvement and de novo genome assembly.
高通量测序技术,也被称为下一代测序(NGS),已经将生物医学研究从遗传学转变为发育生物学。
在短时间内产生大量测序reads的能力,使基因组组装、基因分型、表达谱分析和DNA结合位点的系统识别成为可能,而这在其他方面是困难的或不切实际的。
与所有NGS平台相关的一个关键问题是在read通量和准确性之上的读取长度。
大多数NGS平台产生的相对较短的测序序列长度限制了它们的使用,尤其是在基因组组装和整理方面。
例如,当短序列与参考基因组或彼此之间进行映射时,歧义常常会存在,而读取序列的准确性又会使这种情况进一步复杂化。
短的长度也使它在变异调用和重新基因组组装上有问题。
尽管大量的努力已经在过去的十年里增加阅读的长度,例如,从22日英国石油公司Illumina公司platform2高达300个基点,这些长度仍不满意对于许多应用程序,包括新创基因组装配,完成基因组差距和识别复杂的结构基因组草案的变化。
因此,必须在不同的NGS平台之间进行权衡,以平衡读取长度和产量。
例如,454平台可以产生长达1 Kbp的读值,这在解决基因组缺口时很有用。
这种权衡也催化了第三代测序,这是一种能够产生异常长度序列的测序方法。
PacBio的单分子实时测序(SMRT)就是一个例子,它能够产生高达30 Kbp的测序reads,并被证明在解决复杂的基因组区域4中是有用的。
然而,大多数单次读取的误差率高达15-18% - 5,因此需要在用于基因组组装和其他应用之前进行修正。
indel(插入和删除)的高假阳性率也阻碍了变异调用5中PacBio读取的使用。
Illumina公司最近发布了合成长读技术(http://www.illumina.com/products/truseq- synthet-long -read-kit.ilmn),该技术可以从其现有HiSeq平台生成的短序列中构建合成长序列。
一个令人惊讶的长读取长度加上它的高精度是构成影响从头基因组组装或间隙整理草案基因组。
这项技术也被称为分子测序技术,通过对一种star ascidian动物Botryllus schlosseri进行从头基因组组装和对人类进行全基因组单倍型分析,已经证明了其应用价值。
长读取也被用来解决果蝇基因组中高度重复转座子的子集黑腹果蝇8。
然而,read在解决重复序列和识别复杂基因组变异方面的能力的系统评估仍有待观察。
特别是基因组整理和基因组重新组装中reads的不足还没有被发现。
为此,需要高质量的完整基因组,其中包含所有已解决的重复序列。
秀丽隐杆线虫的基因组是这项任务的选择,因为它有以下特点。
首先,它的基因组是一个没有gap9的完成的基因组,这为阅读准确性和基因组覆盖率的公正评估提供了机会。
其次,所有类型的重复序列都已经明确地解决了,允许在恢复各种重复时系统地评估长读取。
第三,一个注释良好的秀丽隐杆线虫基因组单独与大量的NGS测序数据允许验证潜在的错误,无论是在当前的基因组组装或在长阅读。
最后,同基因的秀丽隐杆线虫基因组很少存在杂合性,这通常是异种杂交生物基因组的从头组装和变异的问题。
通过将长链及其组装的片段映射回秀丽隐杆线虫的基因组,我们系统地描述了基因组改进和重新组装过程中读取序列的前景和不足。
Results
Read accuracy across its length
Given that the synthetic long reads (hereafter referred to as long reads) were assembled from the short reads generated with Illumina HiSeq, only the reads longer than 1500 bp in sizes were retained for all the analysis. A total of 373,121 long reads were generated with two libraries, which covered approximately 24×of C. elegans genome. To filter out the possible contaminations of sequence, all reads were aligned against the NCBI non-redundant genome database by BLASTN. 195 reads were found to be contaminated ones (See Methods) and excluded from further analysis (Supplementary Table S1; Supplementary Figure S1).
To evaluate the sequence accuracy of the long reads, we mapped all the decontaminated reads against the reference genome and calculated the ratio of sequence variations. We defined the variation as a single nucleotide variation (SNV), an insertion or deletion (indel) in the long reads relative to the reference genome. The overall ratios of SNV, insertion and deletion out of the total sequences concerned are 0.011%, 0.019% and 0.101%, respectively, indicating a high degree of sequence accuracy in the long reads. The ratios may be overestimated if they all are treated as read errors because the ratios represent the combined error rate from both reads and the reference genome. For example, a newly acquired variation through spontaneous mutation in the sequenced strain will contribute to the reference error. Notably, the ratio of SNV is much lower than the error rate of Illumina raw reads (~0.6%)10,11, which is likely due to the fact that the long-reads were built from the consensus sequence of the underlying Illumina HiSeq reads that were subjected to self-correction during pre-assembly of the long reads. SNVs and indels that are small in sizes (<9 bps, a default cutoff set by CLC Genomic Workbench) were enriched in both ends of the reads, consistent with a lower quality score in the sequences of these regions (Fig. 1).This could be due to adapter trimming or a fewer number of short reads used to build the consensus sequence at the end of long reads than that used for the central region. Most of the identified indels are relatively short in size (<9 bp) (Fig. 1C,D). However, the density of long indels (> = 9 bp) appears to be constant across read length (Fig. 1E,F). Surprisingly, the deletion rate was observed at a much higher frequency than that for SNV and insertion. All kinds of variation are relatively enriched in the repetitive regions, but depleted in the conserved regions (data not shown).read长度的准确性
考虑到合成长读(以下简称长读)是由Illumina HiSeq生成的短读组装而成,因此在所有分析中只保留长度超过1500bp的读读。
两个文库共生成了373,121个长序列,涵盖了秀丽隐杆线虫约24个基因组。
为了过滤序列中可能存在的污染,BLASTN将所有reads与NCBI非冗余基因组数据库进行比对。
195个reads被发现为被污染的reads(见方法),被排除在进一步分析之外(补充表S1;
补充图S1)。
为了评估长序列的序列准确性,我们将所有去污染的序列与参考基因组进行映射,并计算序列变异的比例。
我们将变异定义为相对于参考基因组的单核苷酸变异(SNV)、长序列中的插入或缺失(indel)。
SNV、插入和缺失占总序列的比例分别为0.011%、0.019%和0.101%,说明在长序列中序列的准确性较高。
如果它们都被当作读取错误来处理,这些比率可能会被高估,因为这些比率代表读取和参考基因组的综合错误率。
例如,在测序菌株中通过自发突变而获得的新变异将导致参考误差。
值得注意的是,SNV的比例远低于Illumina公司生读的错误率(~ 0.6%)10、11,这可能是由于这一事实long-reads建成从底层Illumina公司的共有序列HiSeq读取过程中进行自我修正预装的读取。
SNVs和indels小尺寸(& lt; 9个基点,默认设定的截止CLC基因组工作台)在读取的两端丰富,符合质量分数较低的序列这些区域(图1)。这可能是由于适配器修剪或更少数量的短的读取用于构建共识序列的末尾长读比用于中部地区。
大多数已识别的indels相对较短(9 bp)(图1C、D)。
然而,长indels的密度(>
= 9 bp)在整个读取长度上是恒定的(图1E,F)。
令人惊讶的是,我们观察到的缺失率比SNV和插入的频率高得多。
各种变异在重复区域相对丰富,而在保守区域则相对贫乏(数据未显示)。
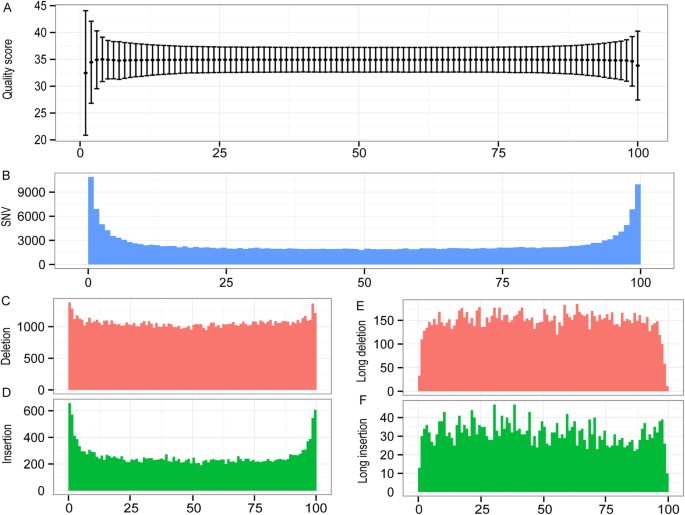
Read quality and mappability across its lengths.
(A) Read quality score (mean ± SD).
(B) SNV count. (C and D) Count of deletion (red) or insertion (green) respectively. (E and F)
Count of long deletion (red) or insertion (> = 9 bp) (green) respectively (see Methods).
Scale in horizontal axis was normalized to a 100% of the read length.
The window size of the plot is 1% of the read length.
Characteristics of read length and its impact on read mappability
To examine whether the reads of different sizes show differential coverage of the C. elegans genome, all the decontaminated long reads were split into 139 subsets with an increment of 100 bp except for those longer than 15.4 Kbp that were merged into the last subset. Reads in all the subsets were subsequently mapped against the C. elegans genome. Median and N50 length of the reads are 6371 and 9175 bp respectively (Fig. 2A,B) with the maximum length of 23 Kbp (data not shown). Plotting of the count versus the size of the long reads demonstrated two peaks with one centering around 1.5 Kbp and the other around 10 Kbp (Fig. 2A). Reads of around 10 Kbp suggest a full reconstruction of the DNA insert inside the library (note the insert size for the long read library is approximately 10 Kbp, see Methods). Though most reads were pre-assembled with a remarkable length of around 10 Kbp, a considerable number of shorter reads existed for which the yields and genomic coverage showed uniform distribution from 1.5 ~ 8 Kbp (Fig. 2B,C).
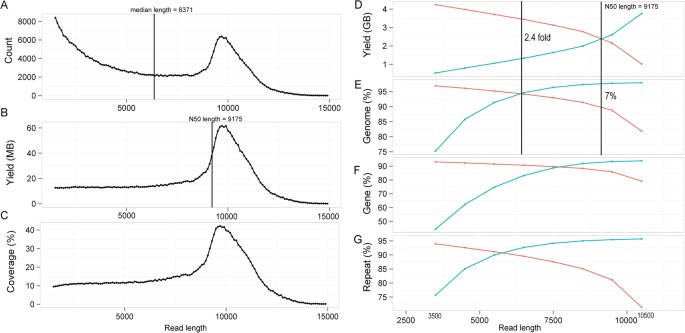
Read count and mappability across its lengths.
(A, B and C) Count, yield and genomic coverage of long read across its lengths respectively with a window size of 100 bp. (D, E, F and G) Accumulative read yield and its coverage of genome, protein-coding genes and repetitive sequences across read lengths respectively. Blue and red lines represent accumulative yield or percentage respectively using the reads shorter or longer than a given read length.
To evaluate the accumulative genomic coverage by the reads of a given length, reads shorter or longer than this length was pooled and mapped back to the reference genome (Figs. 2D to 2G). The accumulative reads shorter than 10.5 Kbp cover most of the reference genome, indicating that the sequences of reads longer than 10.5 Kbp are mainly redundant with those of the shorter reads. The N50 length is the demarcation that the summed yields of all reads longer and shorter than this length are equal to each other. Intriguingly, the reads shorter than the N50 cover about 7% more genome than those longer than the N50 (Fig. 2D, E). The reads shorter or longer than 6.5 Kbp produced the equal mapping coverage (~94%) of the genome (Fig. 2E), whereas the sequence yield of the reads longer than 6.5 Kbp was 2.4 times of that for the reads shorter than this size, indicating that shorter reads can recover various genomic parts with a higher efficiency. To examine how the short reads can outperform the long ones in recovering genome sequences, we investigated the capacity of the two types of reads in recovering sequences of protein-coding genes and non-coding repetitive regions respectively by their accumulative reads. The results show that accumulative reads shorter or longer than 7.5 Kbp can recover the same portion of (~90%) genes (Fig. 2F) while the intersection point for recovering the same portion of repetitive sequences is around 5.5 Kbp (Fig. 2G), indicating that the reads of smaller sizes are more capable of recovering repetitive sequences than those of bigger sizes.
To further examine the impact of read length on its genomic coverage across chromosome, we plotted the coverage of reads with different lengths across chromosomes. Interestingly, the reads with relatively short length tend to concentrate more on the two arms than the mid-part of the chromosome (Supplementary Figure S2A). Consistent with this, the arm regions of C. elegans autosomes is known to contain a higher density of SNPs and repetitive sequences9, indicating that the shorter reads are more capable of recovering repetitive sequences than the longer reads (Supplementary Figure S2A). Taken together, our data demonstrated that the long reads were able to recover most sequences of the C. elegans genome and those with relatively small sizes were more efficient in recovering repetitive sequences than those with relatively big sizes.
Detailed characterization of the long reads’ capability in recovering various repetitive sequences
One of the major challenges in genome assembly is the presence of many types of repetitive sequences that prevent assembly of a complete genome. To evaluate the capacity of the long reads in recovering the resolved repetitive sequences in the C. elegans genome, the long reads were mapped against multiple types of repetitive sequences as defined by Repeatmasker (http://www.repeatmasker.org) (see Methods).
Surprisingly, over 95% of various repetitive sequences can be recovered by a 24 × coverage of long reads except the satellite, RC (Helitron) and simple repeats, which can still be recovered at a ratio of approximately 87%, 86% and 82% respectively (Fig. 3A). To investigate what types of sequences are responsible for the remaining gaps that bear no coverage by any reads, we examined the sequence content within various gaps especially those that are relatively large in size. Scanning the sequences within the gaps against different type of repetitive sequences revealed that three types of repetitive sequences, satellite, RC and simple repeat, were the most enriched repetitive sequences (Fig. 3B; Supplementary Table S5) within the gaps with the DNA repeat contributing most to the yield (Fig. 3C).
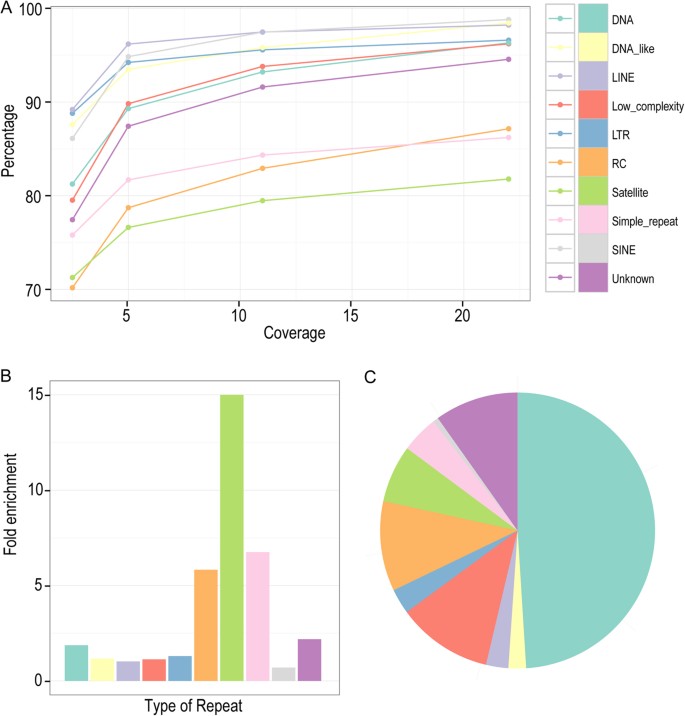
Capacity of long reads in recovering various repetitive sequences.
(A) Effect of read coverage on the recovery rate of various types of repetitive sequence as color coded. (B and C) Relative fold enrichment and composition of various repetitive sequences within the gap regions (the genomic region not covered by any read) respectively. Same color coding scheme is used in (A, B and C).
Consistent with tis, the three types of the most enriched repetitive sequences also showed the lowest recovery rate by the long reads (Fig. 3A). To further understand the likely cause underlying the observed gap region, we first examined the longest gap (~47 Kbp) left by the long reads that is located in the middle of chromosome III (Supplementary Figure S2B, S2C; Supplementary Table S5). A close examination of the sequence content within the gap revealed that the region was mainly comprised of tandem repeat units called satellite repeats “CeRep59”. The highly tandem arrangement of the repetitive sequences possibly prevents the short reads from being placed in this region as described below (Supplementary Figure S2C).
Detailed characterization of the sequence contents within genomic gaps left by the long reads
To examine why the long reads are capable of recovering genomic regions containing certain types repetitive sequences but not those carrying other types of repetitive sequences, we chose to focus on two types of genomic regions both rich in repetitive sequences, one of which was successfully recovered by the long reads (Fig. 4A) while the other failed to be recovered by any reads (Fig. 4B,C). Characterization of sequence contents within these regions will also provide insight into the issues associated with pre-assembly of the long reads to sufficient length. To this end, we first focused on a genomic region of approximately 10 Kbp in size that contains numerous types of annotated repeats, but are fully recovered by the long reads (Fig. 4A). Notably, various types of repeats are present within the region, but they show mixed distribution against one another. In addition, all of the sequence clusters formed by any single repeat-unit are shorter than 500 bp, a size with relatively low copy number that is much smaller than that of the long reads. We next concentrated on another two genomic regions around 10 and 20 Kbp in size respectively because both regions contain a 9-Kbp gap, which bears no coverage by any long read. Examination of their sequence content revealed that the former was almost entirely comprised of a single type of simple repeat (with a repeat unit of “TGATA”) (Fig. 4B) while the latter was composed of nearly all RC repeats, both of which are arranged in tandem (Fig. 4C). It is worth noting that the sizes for many clusters formed by the two types of tandem repeats are bigger than 500 bp, suggesting that it is the tandem arrangement of a single type of repeat sequence forming a cluster that is longer than around 500 bp in size that prevents pre-assembly of a long read with sufficient length (>1.5 Kbp). Therefore, a genomic region rich in these types of repetitive sequences that are arranged in a long tandem stretch (>500 bp) will not be covered by the long reads.

Impact of the arrangement of repetitive sequences in the reference genome on its coverage by the long reads.
(A) An example showing the full recovery of a 10 Kbp genomic region containing various types of repetitive sequences indicated by arrows and differentially color coded by long reads. One satellite repeat within the region is highlighted for its tandem-arranged unit at the bottom. Note, certain reads cannot cover the repetitive region while the others can. (B and C) Examples of the uncovered region (gap) containing simple and RC type of repeat respectively. Note that both repetitive sequences consist of clusters formed by identical tandem units as indicated and the clusters are longer than 500 bp in size while all the repeat clusters in panel (A) are much smaller than this size. Tandem units are highlighted in the bottom. Same color coding scheme is used in (A, B and C). Chromosome coordinates are indicated on the top.
Notably, a single genomic region of roughly 7.2 Kb in size was recovered with unusual depth up to 594×. It is located at the end of chromosome I, within which a single copy of the 18S/5.8S/28S rDNA unit is placed in the C. elegans reference genome (Supplementary Figure S3A). The coverage of synthetic long read is almost uniform with respect to GC-content across the whole genome. Because the raw short reads used to generate the long reads have been normalized before pre-assembly into long read. Only the region with extreme GC content bears lower coverage than average, especially when it contains some tandem repeats. As for the genomic region of 18S/5.8S/28S rDNA unit, with about 47% GC content, the copy number can be estimated roughly by simply dividing the read coverage in this region by that of the genome average. We estimate there are approximate 27 copies of the rDNA gene within the C. elegans genome, which is lower than the previous estimation, i.e., ~55 copies12. Contrary to the rDNA cluster on the chromosome I, the 5S rDNA genic region currently placed on the chromosome V was barely covered by any reads (Supplementary Figure S3B) though about 110 copies of the gene were estimated to be present in the C. elegans genome13. The rDNA genes are expected to be arranged in tandem in the C. elegans genome12,14. It remains possible that the 5S rDNA gene forms extremely tandem repeats that prevents the pre-assembly of the long reads in the relevant genomic regions. Given the capability of the long read to recover the tandem repeats formed by the 18S/5.8S/28S rDNA genes with a repetitive unit of around 7.2 Kbp in size but not those formed by a small-sized repetitive unit, for example satellite, RC or simple repeat and 5S rDNA, the results indicate that the sequence complexity plays a key role in genome recovery rate by the long reads. Sequences with low complexity, for example those composed of short repeats arranged in tandem, will prevent pre-assembly of a long read with the HiSeq raw reads located within the region, leading to a gap left by the long reads. Therefore the genomic regions consisting of tandem repeats with a small-sized repetitive unit are beyond the reach of the long reads. In addition, the coverage of mitochondrial genome by the long reads was relatively low (with only 21 reads mapped) in contrast to the high mitochondrial DNA copy number15, which could be due to the DNA shearing and size selection in long-read library preparation, which demands 10 Kbp in size.
Identification of the relatively long indels within reads
It is surprising that only 710 out of the 11672 SNVs identified with the long reads (Supplementary Table S3) are overlapping with those listed in the SNP database (Wormbase WS242)16, 681 of which are identical between each other. Consistent with this, 140 out of the 681 SNVs are overlapping with those identified using LSJ1 strain, a sister strain of N217, suggesting that these SNVs are possible errors in the reference genome. There are 955 single nucleotide substitution in the C. elegans reference genome, which was annotated as possible reference errors using the existing RNA-seq data derived from mRNA16. 200 out of 955 substitutions are also observed with our SNV data. It has yet to be determined whether the remaining SNVs identified with the long reads represent the errors in reference genome, the read errors or the spontaneous mutations accumulated in our lab strain since its separation with the N2 that was used to produce C. elegans reference genome.
A particular strength of the long reads lies on its capability to identify indels that are relatively big in size, which would be impossible or difficult using the short reads generated with existing NGS platforms. To evaluate the potential of the long reads in recovery of big indels, we first focused on recovery of indels that are longer than 9 bp, (a default cutoff set by CLC Workstation algorithm for variation calling) within individual long reads. We were able to detect 2109 deletions and 274 insertions that were longer than 9 bp within the long reads relative to the reference genome (Fig. 5A; Supplementary Table S4, S5). Because most of the identified indels were supported by multiple independent reads, we decided to examine whether these indels represented errors in C. elegans reference genome, or in the read or a product of spontaneous mutation uniquely accumulated in our N2 strain. To distinguish these possibilities, we performed PCR using the genomic DNAs from the following three sources as a template to validate a subset of the identified indels (see Methods). The genomic DNAs were extracted from our N2 (N2_1), the second N2 that has been separately cultivated from ours for at least 20 years (N2_2) and the Hawaii isolate of C. elegans strain CB4856. To our surprise, 42 out of the 47 tested insertions were confirmed to be present in all three sources whereas only 3, 1 and 1 out of the 17 tested deletions were confirmed to be present in N2_1, N2_2 and CB4856 respectively (Fig. 5E; Supplementary Table S6 ). Presence of insertions in the reads that are confirmed by the PCR product derived from all three sources of C. elegans isolates indicates that the relevant sequences are missing in the current C. elegans genome.
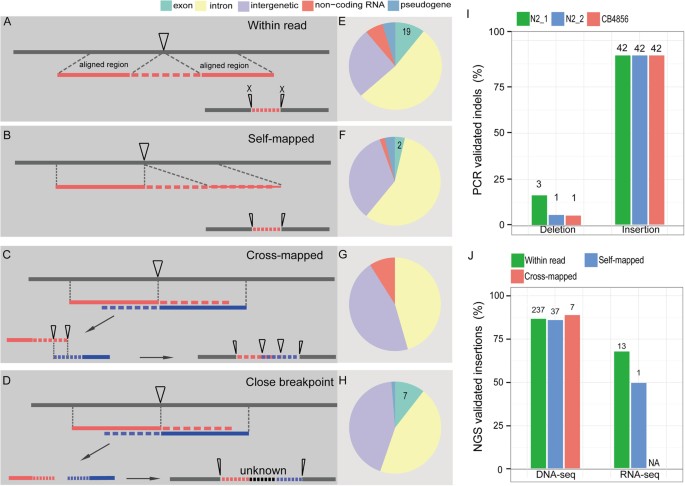
Identification and validation of the long indels (>=9 bp) by PCR and/or NGS data.
(A-D) Shown are the definitions of four possible ways of detecting insertion (insertion position indicated with an inverse triangle). (A) “Within read” type of insertion is defined as an unalignable region within a read against N2 genome while both arms of the read can be successfully aligned with the same reference genome. (B) “Self-mapped” type of insertion is similarly defined as that in (A) except that a second arm can only be aligned with the N2 genome using more permissive alignment parameters (see Methods). (C) “Cross-mapped” type of insertion is defined as an insertion recovered by the overlapping unalignable ends from two independent reads located adjacently in the reference genome. Both unalignable ends that can be partially aligned against N2 genome that are adjacent to each other. (D) “Close breakpoint” type of insertion is similarly defined as that in (C) except that the recovered insertion contains a gap with unknown length due to non-overlapping part between the two unalignable ends. (E-H) Genomic distribution of each type of insertion identified in (A-D). Genomic regions are classified based on their coding potential and differentially colored on the top. Number of insertions located inside coding exon is indicated. (I) Validation results for 51 insertions identified above and 19 deletions identified within long reads by PCR. Shown are the percentages of validated indels that are detected using three different sources of DNA samples as color coded. The numbers of the validated events are indicated above each bar. N2_1 and N2_2 are two N2 C. elegans strain that have been independently maintained over 20 years. CB4856 is the Hawaii isolate of C. elegans. (J) Validation results of three types of insertion as identified in (A, B and C) by NGS data (see Methods). The number of the validated events is indicated above each bar. NA, not applicable.
In contrast, most of the deletions in the reads were not supported by the PCR results. The high false-positive rate of the deletions in reads suggests an intrinsic issue associated with the pre-assembling of the long reads from the raw shorts reads. Consistent with this, most of the deleted sequences within reads tend to contain repetitive sequences (Supplementary Table S3). The elevated false-positive rate of deletion in reads also explains the five-time higher frequency of the observed deletion than that of the insertion and SNV (Supplementary Figure S4). Taken together, it is the presence of certain type of the repetitive sequences especially those positioned in tandem that not only prevents pre-assembly of a long read with sufficient length, but also contributes to the observed higher error rate of deletion in the long reads relative to the reference genome.
Recovery of sequences missing in the current C. elegans genome using the reads with unaligned ends
Given the high degree of accuracy in detecting sequences missing in the reference genome with the long reads, we further explored the possibility of detecting such missing sequences within a single or between two adjacent long read(s) but with a more accommodating alignment method (Figs. 5B,D). Specifically, if a long read can be aligned against the reference genome at one arm but not the other, which is called an unaligned end, the alignment parameters were relaxed so that both arms of the read can be aligned locally against the reference genome except for the mid-part of the read, which is referred to as “Self-mapped” (see Methods). The alignment parameters were relaxed because the initial alignment was made with a high stringency, i.e., demanding 99% identity within a continuous one Kbp region between the query and the reference sequences. To enable the insertions to be recovered by the two adjacent reads that were able to be partially aligned against the reference genome at their one arm but not the other (unaligned end), the possible insertion can be recovered by their overlapping unaligned ends (“Cross-mapped”) (Fig. 5C) or non-overlapping unaligned ends (“Close breakpoint”) (Fig. 5D).
In addition to the 274 insertions (> = 9 bp in size) identified within individual reads using the stringent alignment parameters stated above, another 44, 8 and 75 insertions were recovered with “Self-mapped”, “Cross-mapped” and “Close breakpoint” respectively. A total of 28 identified missing sequences are present within an existing protein-coding exon whereas the remaining ones are located within the intronic or intergenic regions (Fig. 4, Supplementary Table S7).
To validate the identified insertions using independent method, the existing genomic or RNA-seq NGS data were mapped against the identified insertions along with its 2.5-Kbp flanking sequences within the reads (see Methods). Over 85% of the insertions can be confirmed by the genomic sequencing data (Fig. 5F). 14 out of the 21 insertions located within an existing protein-coding exon can be fully or partially validated with the RNA-seq data (Fig. 5; Supplementary Table S7). To illustrate the impact of the insertion on the existing gene model, an insertion located within a protein-coding exon and intron were chosen for gene model repairing respectively (Fig. 6; Supplementary Figure S6). Inclusion of an insertion of 439 bp between position 7485251 and 7485252 on chromosome IV led to a change in the existing gene model of rod-1, i.e., expanding its last exon and splitting it into two separate ones (Fig. 6). The missing fragment was not only confirmed by PCR (Fig. 6B), RNA-seq (Fig. 6C), but also by the multiple alignment of protein sequences among different nematode species (Fig. 6D). Consequently, a total of 129 missing amino acids were recovered relative to the existing ROD-1 protein sequence. Inclusion of another insertion in the first intron of sul-2 between positions of 8158794 and 8158795 on chromosome V produced a new exon, which was confirmed by RNA-seq and multiple alignment of protein sequences from different nematode species (Supplementary Figure S5). As a result, a total of 106 missed amino acids in the current SUL-2 protein sequence can be recovered. At least 40 Kbp missing fragments can be recovered by the long reads and independently confirmed PCR and/or existing NGS data (Supplementary Table S8).
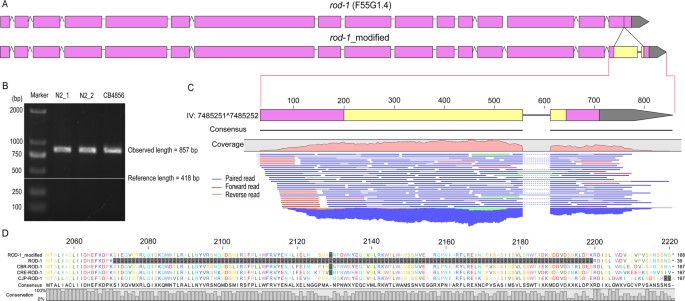
A revised gene model of rod-1 and its validation based on an insertion located within its coding exon.
(A) A revised gene model of rod-1 with an insertion that alters its last exon. Pink, existing exon, yellow, newly added exon. (B) Validation of the insertion by PCR with three sources of DNAs as indicated in 5(I). Shown also are the observed and expected sizes of the PCR products. (C) A magnified view of the affected exon that is confirmed by RNA-seq data whose mapping reads are shown below the corrected gene model. (D) Partial multiple alignment using the C.elegans ROD-1 protein sequence and its orthologs in C. briggsae (CBR), C. remanei (CRE) and C. japonica (CJP). Discrepancies in alignment are shaded in grey.
Usefulness of the long reads in de novo genome assembly
To test the usefulness of the long reads in genome assembly, we performed de novo assembly of C. elegans genome with a 3×, 6×, 12×and 24×coverage of the long reads respectively using two different Overlap Layout Consensus (OLC) based assemblers, Celera and MIRA18. The contigs assembled with Celera appears to be more accurate than those assembled with MIRA in terms of misassembling errors, including relocation, translocation and inversion (Table 1). However, the contigs assembled with MIRA have a longer N50 length while the achieved genome coverage by the assembled contigs is comparable between the two assemblers (Fig. 7; Table 1). Assembly with both algorithms demonstrated that genome coverage by the contigs plateaued with approximately 12×coverage of the reads, which represents approximate 93% of the C. elegans genome (Fig. 7B). However, the N50 length can still benefit substantially from the increasing read coverage from 12×to 24×(Fig. 7A), indicating a higher read coverage is needed for de novo assembly of a genome with a high quality, especially for those genomes that are big in size or contain substantial heterozygosity. Notably, the contigs produced with both assemblers suffer from a relatively high misassembling rate (Table 1).
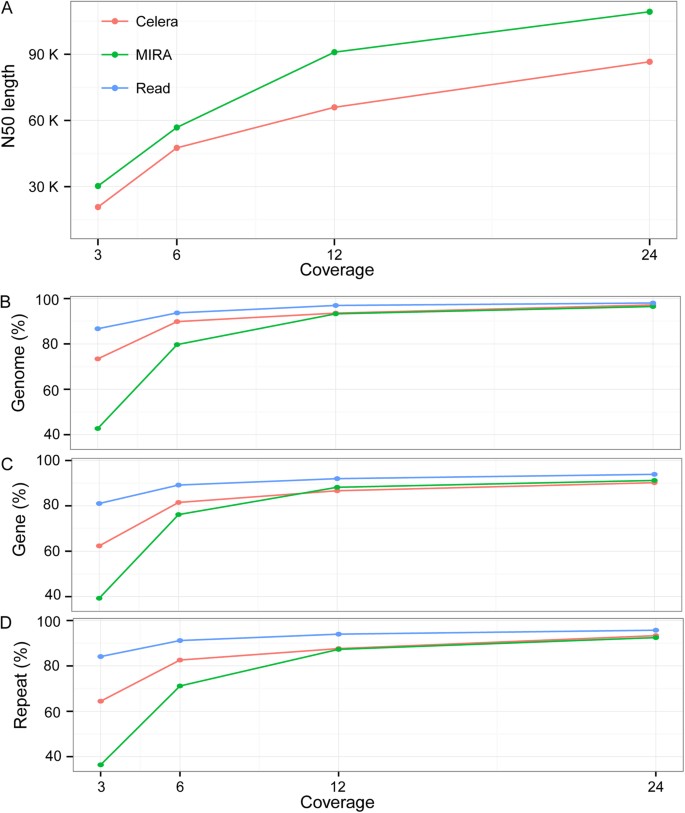
Effect of sequencing depth on de novo genome assembly using two genome assemblers.
(A) Plotting of contig N50 length against the sequencing depth. (B, C and D) Plotting of read (blue) and contig (red and green) coverage of genome, protein-coding genes and repetitive sequences against the sequencing depth respectively. Contigs were assembled using Celera (red) or MIRA (green) assembler with 3×, 6×, 12× and 24× reads.
Given the bias of the read sequence and a relatively small N50 contig size as well as a high rate of misassembling errors associated with the long reads compared with that of other long reads such as PacBio19, complement of Illumina long read sequencing by independent sources of reads, for example from PacBio and/or mate-pair sequencing is necessary to generate a “finished” grade of genome.
Discussion
A finished isogenic genome with resolved repetitive sequences allows an unbiased assessment of read accuracy
The estimated error rate of the Illumina long-reads using Drosophila melanogaster genome8 differs substantially from ours. For example, their SNV rate is about four times higher than ours (0.051% vs 0.011%) while their deletion rates is about one quarter of ours (0.029 vs 0.101%) respectively, though the insertion rates are comparable (0.019% vs 0.017%). One plausible reason underlying the observed discrepancy is that a repeat-masked genome was used for the data analysis in the previous study while an unmasked complete genome containing all types of repetitive sequences was used in this study. For example, our deletion rate in reads is 0.0017% if the mapping was performed with a repeat-masked C. elegans genome (around 14 Mbp masked, see Methods), which is comparable to that reported with D. melanogaster. Inclusion of all types of repetitive sequences allows an unbiased and systematic assessment of the read accuracy and mappability. Another possible reason underlying the elevated SNV rate observed in D. melanogaster genome is likely due to the sequence uncertainties associated with heterozygosity that have been sequenced with a lower confidence20, whereas hermaphroditic mode of reproduction produces an isogenic genome in C. elegans. In addition, a likely higher error rate in D. melanogaster reference genome than that in C. elegans may also contribute to the elevated error rate in the former species.
一个完成的同基因基因组与解决重复序列允许无偏见的评估读的准确性
使用果蝇基因组8对Illumina长读的估计错误率与我们的有本质上的不同。
例如,他们的SNV比率大约是我们的四倍(0.051% vs 0.011%),而他们的缺失比率分别是我们的四分之一(0.029 vs 0.101%),尽管插入比率是可比较的(0.019% vs 0.017%)。
观察到的差异背后的一个可能的原因是,在之前的研究中使用了一个重复掩蔽的基因组进行数据分析,而在本次研究中使用了一个包含所有类型重复序列的未掩蔽的完整基因组。
例如,如果用重复掩蔽的C. elegans基因组(约掩蔽14mbp,参见方法)进行绘制,我们在reads中的缺失率为0.0017%,这与D. melanogaster的报道相当。
包含所有类型的重复序列允许对读取精度和可映射性进行无偏和系统的评估。
在黑线虫基因组中观察到的SNV率升高的另一个可能原因可能是由于与杂合性相关的序列不确定性,而杂合性排序的可信度较低20,而在秀丽隐杆线虫中,雌雄同体的繁殖方式产生了同基因的基因组。
此外,黑腹线虫参考基因组的错误率可能比秀丽隐杆线虫更高,这也可能是前一物种错误率升高的原因。
The long reads hold promise for repairing even the “finished” grade genome
The short reads generated by NGS platforms have been widely used in genomic variation calling between wild isolates of the same species including C. elegans10. They have also been used to detect possible errors in reference genomes. For example, sequencing of an N2 sister strain, LSJ1 with read length of 36 bp has identified an insertion of 34 bp in reads that is mapped to the genomic position of 3568818 and 3568819 of chromosome II17. Alignment of our long reads against the C. elegans genome extends the 34-bp insertion into its full size of 339 bp, demonstrating the potential of the long reads in recovering the genomic fragments that are relatively big in size but missing from the reference genome, which would be difficult with the short reads. We expect the long reads will be useful in refining the draft genomes of other Caenorhabditis species, especially those of C. briggsae and C. nigoni, that have recently been developed as a model for study of speciation genetics21,22.
这些冗长的read有望修复甚至已经完成的基因组
NGS平台产生的短读子被广泛应用于包括秀丽隐杆线虫在内的同一物种野生分离株的基因组变异呼叫。
它们也被用来检测参考基因组中可能的错误。
例如,对一个读长为36 bp的N2姐妹株LSJ1进行测序,在reads中发现了一个34 bp的插入,该插入被定位到II17染色体的3568818和3568819的基因组位置。
对准我们长期的阅读对秀丽隐杆线虫基因组扩展了34个基点插入339个基点的全尺寸,展示的潜力长读入恢复基因片段相对规模大但没有参考基因组,这将是困难的和短的读取。
我们预计,这些长序列将有助于精炼其他Caenorhabditis物种的基因组草案,尤其是C. briggsae和C. nigoni的基因组草案,它们最近已被开发为物种遗传学研究的模型22,22。
Novel assembling algorithm is needed to accommodate the long reads in de novo genome assembly
OLC based assemblers are expected to accommodate the long reads better in de novo genome assembly than the de Bruijn graph-based ones, which is likely due to the limitation on the K-mer size imposed by the latter23. For example, the N50 size for the contigs produced by MIRA and Celera using 24 × reads is 109.3 and 86.6 Kbp respectively without any scaffolding steps (Table 1) while the N50 size is only around 22 Kbp for the contigs produced using de Bruijn graph-based assembler in CLC Genomic WorkBench with the same 24 × reads in C. elegans (our unpublished data). However, the contigs produced by MIRA and Celera still suffer a relatively high mis-assembly rate. The most common errors include relocation and translocation. To explore the potential causes underlying the observed errors, we pulled out a typical translocation event, in which a fragment from chromosome V was incorrectly merged with another one from chromosome I (Supplementary Figure S6A). First, there is an apparent error in pre-assembly of the read that spans the two fragments, which is caused by two 1.2 Kb-repeats that are nearly identical in sequence and located at the opposite ends of each contig (Supplementary Figure S6A). Second, about half of the long reads can still be correctly preassembled so that they are able to span the repetitive regions correctly (Supplementary Figure S6B, S6C). However, Celera assembler tends to neglect these reads that correctly span the boundary between the two contigs but decides to use the single incorrectly assembled read that is substantially longer than the correctly assembled ones to extend the contig. This is because the assembler chooses the read with the longest overlapping region instead of the one with a higher coverage but a shorter overlapping size. This type of error is expected to be fixed by fine-tuning the weighting between read overlapping length and coverage in the assembling algorithm.
为了适应基因组重新组装过程中的长读取,需要新的装配算法
基于OLC的装配器预计比基于de Bruijn图的装配器在重新组装基因组时更好地适应长读,这可能是由于后者对K-mer大小施加的限制23。
例如,将军的大小产生的叠连群米拉和塞莱拉使用24读取分别是109.3和86.6 Kbp没有任何脚手架步骤(表1),而将军大小只有大约22 Kbp的叠连群用de Bruijn基于汇编CLC基因组工作台使用相同的24读入秀丽隐杆线虫(我们的未公开的数据)。
然而,MIRA和Celera生产的contigs仍然存在较高的误组装率。
最常见的错误包括重新定位和易位。
为了探究所观察到的错误背后的潜在原因,我们提取了一个典型的易位事件,其中来自染色体V的一个片段与来自染色体I的另一个片段不正确地合并(补充图S6A)。
首先,跨越两个片段的read在预装配时存在明显的错误,这是由于两个1.2 kb的重复序列几乎相同,并且位于每个contig的两端(补充图S6A)。
其次,大约有一半的长读仍然可以正确地预组装,使它们能够正确地跨越重复区域(补充图S6B, S6C)。
然而,Celera assembler倾向于忽略这些正确跨越两个contig边界的读取,而是决定使用一个错误组装的读取,该read比正确组装的读取长得多,以扩展contig。
这是因为汇编程序选择重叠区域最长的读取,而不是覆盖范围较高但重叠大小较短的读取。
在装配算法中,通过微调读取重叠长度和覆盖率之间的加权来修正这类误差。
In summary, the Illumina synthetic long reads are highly accurate and able to recover most types of repetitive sequences although the reads of different lengths show certain coverage bias against target regions with different sequence contents. The reads would be invaluable in genome finishing, including recovery of missing sequences and/or detection of sequence rearrangement in a draft genome. Tandem arrangement of short repeat unit prevents preassembly of reads with sufficient length using HiSeq raw reads, especially when the region consisting of the tandem repeat is longer than 500 bp, which results in failed recovery of these genomic regions by the long reads. De novo assembly with the long reads demonstrates their use in genome assembly, but also highlights a need of novel genome assembling algorithm to accommodate the long reads.
综上所述,尽管不同长度的reads对不同序列内容的目标区域有一定的覆盖偏差,但是Illumina合成长reads的准确率较高,能够恢复大部分类型的重复序列。
这些读取在基因组整理方面将是无价的,包括恢复缺失序列和/或检测基因组草图中的序列重排。
短重复序列单元的串联排列阻止了HiSeq raw reads预组装足够长的reads,特别是串联重复序列组成的区域超过500 bp时,导致这些基因组区域在长读时无法恢复。
长读序列的从头组装展示了它们在基因组组装中的应用,但也强调了需要新的基因组组装算法来适应长读序列。
Methods
C. elegans strains
N2 strain from our own lab (shipped from Seattle, WA, USA in 2010) was used for extraction of genomic DNA and sequencing library preparation. For PCR validation of the detected indels, genomic DNAs from three different lines of C. elegans were used as a template, including our own N2 strain (N2_1), another N2 strain that has been independently cultivated for at least 20 years from our N2 strain obtained from a local lab (N2_2) and a Hawaii isolate of CB4856.
Library preparation, sequencing and read pre-assembly
The libraries were prepared according to Illumina’s protocol. Briefly, 500 ng high quality gDNA was sheared by g-Tube (Covaris). The sheared gDNA went through end repair, dA-tailing and adaptor ligation. The ligated products were size-selected for 8–10 Kbp range by agarose gel and eluted followed by quantification with qPCR. Based on qPCR quantification, the long insert library was diluted and distributed into a 384-well plate, with each well containing about 3 fg of the library. After long range PCR, the PCR product went through Nextera tagmentation and barcoding with 384 different barcoding PCR primers. The final products were pooled together, cleaned, concentrated and size-selected. A total of two final libraries were made using the same long insert library mentioned above. Each library was sequenced by a HiSeq 2500 Rapid Run Flowcell at 2 × 101 bp with 8-bp barcode. The primary sequencing results were streamed to BaseSpace and pre-assembled using the TruSeq Long-Read Assembly App v1.0 (https://basespace.illumina.com/). The application first pre-processed the reads to correct the sequencing and PCR errors and normalized read depths across fragments. In the second stage, a string graph was constructed using the String Graph Assembler (SGA); the resulting graph was then cleaned using the paired-end information from the short reads to produce an initial set of contigs. In the third stage, the contigs were subjected to further scaffolding to resolve repeats and fill the gaps that were created due to low sequencing coverage. In the final stage, the scaffolds were examined for possible sequence errors and mis-assemblies and corrected. The assembled long-reads were generated as FASTQ files that were used in the subsequent analysis.
Read decontamination and mapping
To filter out the possible DNA contaminations from other organisms in the reads, BLASTN was run locally against NR database with default parameters using all reads as an input to identify the potential contaminations. The hits with best match (smallest e-value) to a target that does not belong to C. elegans genome were treated as contaminated reads and thus removed from the downstream analysis (Supplementary Figure S1).
To map the reads against reference genome, C.elegans genome was downloaded from WormBase (WS242)16. All the reads bigger than 1.5 Kbp in size were mapped against the reference genome using BWA-MEM using the following parameters, i.e., single-ended, minimum seed length of 1000 bp and band width of 23 Kbp (the longest size of the reads)24. Approximately 0.07% (around three thousands) of the reads cannot align with the reference by BWA-MEM using the above parameters. To accommodate these reads, they were mapped again to the genome as stated above except with a minimum seed length as default. The resulting BAM files from two rounds of mapping were merged, sorted and indexed with SAMtools25 and the read coverage among chromosomes were calculated using BEDTools (v2.20.1)26. Given the significance of mapping method in variation finding, for example, mapping of the long reads using small seed length will generate small mapping segments and make mapping ambiguous, we employed 2 rounds of BWA-MEM mapping with different band size respectively to minimize the ambiguous mapping segments.
The C. elegans repetitive sequences were identified using the existing ones detected by RepeatMasker (http://www.repeatmasker.org), which defined 10.31% and 2.28% of the C. elegans genome as interspersed and locally repetitive sequences. The former can be further divided into seven main classes, including DNA repeat, LINE, SINE, LTR, RC/Helitron, DNA like repeat and some unknown repeats. The latter can be further divided into simple repeat, satellite repeat and low complexity region. The gene and tandem repeat annotations were retrieved from WormBase16. Only annotations for 20487 protein-coding genes were used for analyzing the gene coverage. A repeat or a gene was defined as “recovered” by the long reads when the full length of its sequence is covered at least once by a single or overlapping read(s).
Variation calling
The positions of SNVs and indels identified within reads relative to the reference genome were output by parsing the CIGAR string and MD tag from the mapping BAM files. To characterize the genomic distribution of the variations between the long-read sequences and the reference genome, the mapping BAM files were parsed using “probabilistic variation detection” tool in CLC Genomic Workbench 7.0.3, with the following parameters: the minimum coverage of 1, minimal variation count of 1, probability larger than 0.5 and ploidy of 2 (http://www.clcbio.com/support/white-papers/). The resulting variations in the long reads relative to the reference genome were classified into four categories, namely SNV, insertion, deletion and complex variation. SNV was defined as a single nucleotide variation, meaning one base was substituted by another. “Insertion” was defined as an inserted sequence in reads relative to the reference genome, while the “deletion” as a deleted sequence in the long read relative to the reference genome.
The “probabilistic variation detection” tool of CLC Genomic Workbench can only recover insertion sequence within a single read. To recover the possible insertions that may be resolved by two long reads located adjacent to each other in the reference genome, the unaligned ends generated by the read mapping were parsed using the “indel and structure variation finding” tool in CLC Genomic Workbench. The “indel and structure variation finding” tool was used to locate the breakpoint of the alignment and the identified unaligned end was mapped back to the reference genome locally. Alternatively, the local unaligned ends generated by multiple independent reads that were closest between each other in distance were mutually aligned. Using the two tools, four types of insertions can be recovered, i.e., the “Within read”, “Self-mapped”, “Cross-mapped” and “Close breakpoint”. Identical insertions recovered with two independent methods were merged to a single one.
PCR validation of the identified indel that is longer than 40 bp in size
To validate the identified long insertions and deletions within or between long reads, a subset of 48 insertions and 17 deletions that are longer than 40 bp were validated using PCR (Supplementary Table S6). The presence or absence of an indel can be distinguished by examining the product size.
Validation of the identified insertions using existing NGS data
Two datasets, DRR008443 and SRR1050780 were downloaded from Sequence Read Archive (SRA)27 to validate the insertions that are longer than nine bp in size. The former is the 70 × N2 genomic DNA sequencing data generated with HiSeq (paired-end, 2 × 100 bp, library size of 500 bp). The latter is the RNA-seq data of mix-staged N2 mRNA (paired-end, 2 × 75 bp, library size of 300 bp). All insertions along with its two flanking 2500 bp sequences were pooled into a single file to be used as a reference. The genomic DNA sequencing data were mapped against the reference with “mapping reads to reference” tool of CLC Genomic Workbench using default settings. If an insertion was fully covered at least 10×across both boundaries by the paired end reads, the insertion was marked as “confirmed”. RNA-seq data were mapped against the reference using “large gap read mapping” tool of CLC Genomic Workbench to validate the insertion located within an existing protein-coding gene model. The region with at least 2×coverage was treated as an exonic region. The gene model was re-annotated/repaired using the “gene prediction tool”. The protein coding sequences of sul-2 and rod-1 and their homologous proteins were downloaded from Wormbase16. Multiple sequence alignment was carried out with the CLC genomic workbench using the default settings.
De novo genome assembly and contig assessment
The subset of reads were generated randomly from the combined FASTQ file of the two libraries containing 24×reads (length > 1.5 Kb) to give rise to 3×, 6×and 12×of long reads. Two overlap-layout-consensus (OLC) assemblers were used to assemble the genome region respectively. The major parameters for Celera (8.1)28 were ovlMinLen = 500, unitigger = bogart, ovlMerSize = 31, utgGraphErrorRate = 0.0045, utgMergeErrorRate = 0.003, merThreshold = auto*2. And the major parameter for MIRA (4.0.1)18 were -AL:mo = 500, -SK:bph = 31, -SK:pr = 99, -AL:mrs = 99, -CO:fnicpst = yes. To evaluate the characteristics of the contigs assembled with Celera (8.1) or MIRA (4.0.1), the prevalence of SNVs, indels and other mis-assembly events were calculated using the QUAST (2.3)29.
Data availability
Sequence data can be found under the NCBI BioProject: SRP047457, BioSample: SRS708640 and Experiment: SRX709602. (SRA RUN No pending).