RNA测序相对基因表达芯片有什么优势?
RNA-Seq和基因表达芯片相比,哪种方法更有优势?关键看适用不适用。那么RNA-Seq适用哪些研究方向?是否您的研究?来跟随本文了解一下RNA测序相对基因表达芯片有什么优势?
无假设的研究设计和更高的发现能力
RNA-Seq是一种基于测序的强大方法,让研究人员能够打破传统技术的低效和花费,如实时定量PCR(RT-PCR)和芯片。无论是将RNA-Seq添加到现有的研究方法中,还是从一种方法彻底转换到另一种,RNA-Seq都带来了许多显而易见的优势。这种方法不需要预先设计的探针,因此数据集是无偏倚的,实现了无假设的实验设计2,3。这种类型的NGS分析对转录本和变异发现研究而言是一种有力工具,而传统的芯片方法无法实现。
更宽的动态范围和更高的灵敏度
芯片测定连续探针强度,而RNA-Seq不同,它对与参考序列比对的单条序列进行定量,产生了离散的(数字)读数2。此外,通过增加或减少测序读数(覆盖水平或覆盖深度),研究人员可以微调实验的灵敏度,以适应不同的研究目标。这个过程的数字化属性以及控制覆盖水平的能力支持一个非常宽的动态范围,提供绝对而不是相对的表达值1-3。假设有1000-5000万条定位读数,则RNA-Seq的动态范围可跨越5个数量级(>105),这通常比大多数芯片技术(103)高出几个数量级2,4。因此,研究表明,RNA-Seq可检测的差异表达基因比例比表达芯片更高,特别是低丰度的基因4,5。
检测选择性剪接位点和新型异构体以及非编码RNA的能力
除了基因表达谱分析,RNA-Seq还能鉴定选择性剪接异构体、剪接位点和等位基因特异的表达–所有这些都在单个实验中完成1,2。此外,RNA-Seq还能测序极短的片段,且文库制备方法可包含或排除mRNA提取,这样它就能检测并测序小RNA及多种形式的非编码RNA,如小干扰RNA(siRNA)、microRNA(miRNA)、核仁小RNA(snoRNA)及转运RNA(tRNA)1,2。测序小片段的能力也让降解的RNA样品产生高质量的数据,如福尔马林固定、石蜡包埋(FFPE)样品6。随着转录组的新特征被不断发现,测序数据还可以重新分析。对于芯片,假设原始样品仍然可用,样品也必须重新走完整个芯片流程,从探针设计到实验室工作7。总之,RNA-Seq相对于芯片而言具有很多优势(表1)。它带来转录组范围的覆盖、宽的动态范围以及高灵敏度的独特组合,让研究人员能够研究和了解正常发育和疾病的分子机制。
表1 RNA-Seq技术与表达芯片的比较!--?
|
应用 |
RNA-Seq |
芯片 |
|
每次运行之间的重复性高 |
是 |
是 |
|
动态范围与细胞内真正的转录本丰度相当 |
是 |
否 |
|
能够检测选择性剪接位点和新的异构体 |
是 |
否 |
|
无参考基因组的de novo分析 |
是 |
否 |
|
再次分析数据 |
是 |
否 |
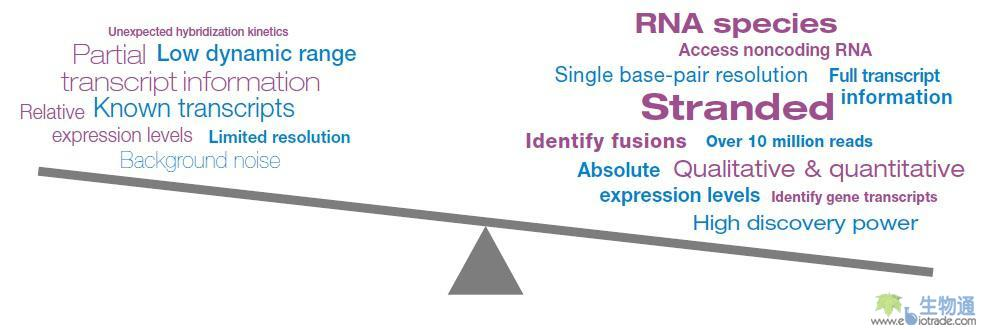
总结:RNA-Seq相对基因表达芯片的优势(图1)
• 在转录本水平提供灵敏且准确的基因表达测定
• 产生定性且定量的数据
• 捕获剪接点、融合、编码及多种形式的非编码RNA,如siRNA、miRNA、snoRNA和tRNA
• 覆盖非常宽的动态范围
• 在降解RNA上表现出色,如FFPE组织样品
• 在数据中维持和追踪链特异的信息
• 为发现型的应用带来一种更强大的方法
• 在大型研究和大量样品时可扩展
有了这些优势,RNA-Seq正在推动研究的步伐,在多个领域催生高影响力的论文,包括癌症研究、复杂疾病和病毒学(图2)
RNA-Seq适用哪些研究方向?
转化医学--癌症研究
通过RNA测序 (RNA-Seq)来监控癌症基因表达和转录组改变,能帮助回答疾病分类和进展等研究问题。癌症积累了大量的遗传改变,但通常只有少数改变能真正推动肿瘤发展。
转化医学--复杂疾病研究
基因表达的差异与个体间的表型变异相关联。表达数量性状位点 (eQTL) 调控mRNA表达水平,让研究人员能有效地将表达水平定位到个体之间基因组中的差异。
基础研究--农业基因组学
RNA测序正在彻底改变动植物中基因表达的探索,为发育、疾病和压力状态下的表达水平改变提供新的见解。它可用于阐明基因和蛋白的功能及相互作用,鉴定动物或植物基因组所产生的组织特异的RNA转录本列表(mRNA、非编码RNA和小RNA),以及SNP发现。
基础研究--细胞生物学
核糖体轮廓分析是一种研究翻译调控(即基因表达的调控)的强大技术。这一应用通过估计蛋白质丰度和翻译调控,在基因组学/转录组与蛋白质组学之间架起桥梁,从而增加了从RNA-Seq中获得的mRNA丰度信息。
1. Ozsolak F, Milos PM. RNA‑Sequencing: advances, challenges and opportunities. Nat Rev Genet. 2011;12:87‑98.
2. Wang Z, Gerstein M, Snyder M. RNA‑Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57‑63.
3. Wilhelm BT, Landry JR. RNA‑Seq—quantitative measurement of expression through massively parallel
RNA‑Sequencing. Methods. 2009;48:249‑57.
4. Zhao S, Fung-Leung WP, Bittner A, Ngo K, Liu X. Comparison of RNA‑Seq and microarray in transcriptome profiling of activated T cells. PLoS One. 2014;16;9(1):e78644.
5. Wang C, Gong B, Bushel PR, et al. The concordance between RNA‑Seq and microarray data depends on
chemical treatment and transcript abundance. Nat Biotechnol. 2014;32:926-932.
6. Illumina (2014) TruSeq RNA Access Library Prep Kit Data Sheet. (www.illumina.com/content/dam/illumina-marketing/documents/products/datasheets/datasheet-truseq-rnaaccess.pdf).
7. Illumina (2011) RNA‑Seq Data Comparison with Gene Expression Microarrays White Paper (www.europeanpharmaceuticalreview.com/wp-content/uploads/Illumina_whitepaper.pdf).
8. Grant award data obtained from the NIH website (projectreporter.nih.gov). Accessed March 2016.