1 预备知识
1.1 分子基础概念
1.1.1 质谱学
识别给定样本中存在的分子是相当具有挑战性的。目前最流行的技术是质谱分析法。质谱分析的基本思想是用电子轰击样品。这种轰击把多个分子粉碎成碎片。这些碎片通常会电离,即获得或失去电子从而带电。检测到的带电碎片的扩散称为光谱(spectrum)
许多研究人员正在积极研究利用深度学习算法改进质谱分析技术,以便从检测到的带电光谱中轻松识别原始分子。
1.1.2 分子键
分子键有很多种类型,包括共价键和集种非共价键。
共价键实涉及两个原子之间共享电子,是最强的化学键。
非共价键不涉及原子间电子的直接共享。非共价键包括氢键、盐桥、Π堆积等。这些类型的相互作用通常在药物设计中扮演着重要角色,因为大多数药物通过非共价键相互作用于人体中的生物分子。
1.1.3 分子构型
分子图描述了分子中的一组原子以及它们是如何键合在一起的。而原子在3D空间中是如何相对放置的,这叫作分子构型。
1.1.4 分子手性
有些分子(包括许多药物)有两种形式,它们互为镜像,这叫作手性(chirality)。
手性分子同时具有“右手性”形式(也称为“R”型)和“左手性”形式(也称为“S”型),如图所示。
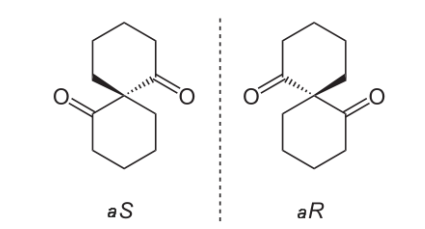
一种药物的两种手性形式有可能与完全不同的蛋白质结合,并对身体产生非常不同的影响。在许多情况下,只有一种形式的药物具有预期的治疗效果,另一种形式只产生额外的副作用,没有任何好处。
1.2 分子的特征表示
为了对分子进行机器学习,需要把它们转换成特征向量,作为模型的输入。
1.2.1 SMILES字符串和RDKit
SMILES是一种用文本字符串定义分子的常用方法,这个首字母缩略词代表“简化的分子输入线输入系统(SimplifiedMolecular-InputLine-Entry System)”。SMILES字符串以对化学家来说既简洁又直观的方式描述了分子的原子和键。对于非化学家来说,这些字符串看起来像是毫无意义的随机字符模式。例如,OCCclc(C)In+(cs)Cc2cnc(C)ncN”描述了重要的营养物质硫胺素,也称为维生素B1。
一些深度学习模型直接接受SMILES字符串作为输入,试图学习识别文本表示中的有意义的特性。
但更常见的情况是先将字符串转换为更适合当前问题的另一种表示形式(或特征化)。
RDKit提供了许多用于处理SMILES字符串的功能,在将数据集中的字符串转换为分子图和下面描述的其他表示形式方面发挥着核心作用。
1.2.2 扩展-连接指纹
化学指纹是由1和0组成的向量,表示分子中特定特征的存在或不存在。扩展连接指纹(ECFP)是一类结合了几个有用特性的特征化表示它们可将任意大小的分子转换成固定长度的向量。
指纹向量的每个元素都表示存在或不存在由某些局部原子排列所定义的特定分子特征。分子指纹算法首先独立地考虑每个原子,并观察原子的一些特它的元素、形成的共价键的数量等。这些属性的每个唯一组合都是一个特征,向量的对应元素被设置为1来表示它们的存在。然后,算法向外扩展将每个原子与它所连接的所有原子结合起来,这定义了一组新的更大的特征并设置了向量的相应元素。这种技术最常见的变体是ECFP4算法,它允许子片段在中心原子周围有两个键的半径。
ECFPS有一个重要的缺点:指纹编码了分子的大量信息,但有些信息确实丢失了。两个不同的分子有可能拥有相同的指纹,并且给定一个指纹,不能唯地确定它来自哪个分子。
1.2.3 分子描述符
另一种观点认为,用一组理化描述符来描述分子很有效。这些通常对应于描述分子结构的各种计算量。这些量,如对数分配系数或极坐标表面积,通常是由经典物理或化学推导出来的。
这种特征化方式对于某些问题显然比其他特征表示更有用。它最适合那些比较依赖于分子一般性质的预测,而不太可能用于预测依赖于原子详细排列的性质。
1.2.4 图卷积
上文描述的特征表示方法是由人类设计的,考虑了如何用一种可以作为机器学习模型输入的方式来表示分子,然后手动编码表示。
能不能让模型自己找出表示分子的最好方法?毕竟,这就是机器学习的全部内容:可以尝试从数据中自动学习,而不是自己设计一种特征。
作为类比,考虑用于图像识别的卷积神经网络。卷积神经网络的输入是原始图像,它由表示了每个像素的数字向量组成,例如三个颜色分量构成的数字向量。这是一个非常简单、完全通用的图像表示。第一个卷积层学习识别简单图案,如垂直或水平线,它的输出仍然是表示了每个像素的数字向量,但现在它以更抽象的方式表示,每一个数都表示局部几何特征的存在网络通过一系列层继续运行。每一层都输出一个新的图像表示,它比前一层的表示更抽象,并且与原始颜色分量的关联性较小。这些表示是从数据中自动学习的,而不是由人类设计的。没有人告诉模型寻找什么模式来识别图像中是否包含一只猫,这个模型通过训练自己算出来。
图卷积网络(Graphconvolutionalnetworks)采用了同样的思想,并将其应用于图形输入。就像普通的CNN从表示了每个像素的数字向量开始计算一样,图卷积网络也以表示了每个节点和/或边的数字向量开始计算。当用图形表示个分子时,这些数字可以是每个原子的高级化学性质表示,比如原子的元素电荷和反应状态。就像普通卷积层根据输入的局部区域为每个像素计算一个新向量一样,图卷积层为每个节点和/或边计算一个新向量。通过将一个学习过的卷积核应用到图的毎个局部区域来计算输出,其中“局部”是根据节点之间的边来定义的。例如,图卷积层可以基于原子和与它直接相连的任何其他原子的输入向量,为每个原子计算一个输出向量。
图卷积网络是分析分子的有力工具,但它们有一个重要的局限性:计算完全基于分子图。由于没有用分子构型的任何信息,图卷积网络不能预测任何与构型相关的东西。这使得图卷积网络最适合于小的且大都是刚性的分子。
1.3 蛋白质
1.3.1 蛋白质结构
蛋白质是完成细胞中大部分工作的微型机器,尽管它们的体积很小,但也很复杂。典型的蛋白质是由成千上万个原子以精确的方式排列而成的要了解任何一台机器,你必须知道它是由什么部件组成的,以及这些部件是如何组合在一起的。你想了解一辆汽车,需要知道它的底部有轮子,中间有空的空间来容纳乘客,以及乘客可以通过门进出。
蛋白质也是如此,要了解它是如何工作的,必须确切地知道它是如何被组合在一起的,此外需要知道它是如何与其他分子相互作用。很少有机器是单独工作的。汽车与它所载的乘客、它所行驶的道路以及允许它移动的能源相互作用这也适用于大多数蛋白质,它们作用于其他分子(例如,用于催化化学反应),被其他分子利用(例如,调节它们的活动),并从其他分子中汲取能量。所有这些相互作用都取决于两个分子中原子的具体位置,要理解它们,必须知道原子在3D空间中是如何排列的。
目前有三种方法来确定蛋白质的结构,包括射线晶体学、核磁共振(简称NMR)和低温电子显微镜(简称cryo-EM)。
预测蛋白质结构的方法主要有两种:
第一种称为同源建模。蛋白质序列和结构是几十亿年进化的产物。如果两种蛋白质是最近才彼此分离的近亲(技术术语是“同系物”),它们可能有相似的结构。要通过同源建模来预测蛋白质的结构,首先要寻找结构已知的同源物,然后根据两种蛋白质序列之间的差异来调整它。同源建模在确定蛋白质的整体形状方面效果相当好,但它往往会得到错误的细节。当然,这需要你已经知道同源蛋白质的结构。
另一个主要的方法是物理建模。利用物理学定律的知识,探索蛋白质可能呈现的许多不同构象,并预测哪一种构象最稳定。这种方法需要大量的计算时,直到大约十年前,这根本是不可能的。即使在今天,它也只适用于小的快速折叠的蛋白质。而且,它需要物理近似来加速计算,而这些近似会降低计算结果的精度。物理建模通常会预测出正确的结构,但并不总是如此。
2 基于深度学习的生命科学
2.1 基于图卷积神经网络的分子溶解度预测问题
2.2 类药物小分子与蛋白质结合问题
这个问题是药物发现的根本问题。以一种针对性方式调节单个蛋白质通常会产生显著的治疗效果。例如,突破性的癌症药物伊马替尼与BCR-ABL紧密结合,这是其疗效的部分原因。对于其他疾病,寻找具有相同功效的单一蛋白质靶可能具有挑战性,但是这种想法仍然有用。
将针对某种疾病的药物设计问题降为设计一种与给定蛋白质紧密结合的药物的问题非常有用。但必须认识到,实际上,任何一种药物都会与体内许多不同的子系统相互作用。
2.3 转录因子的结合问题
作为将深度学习应用于基因组学的一个例子,考虑预测转录因子结合的问题:TFs是与DNA结合的蛋白质。当它们结合时,会影响附近基因被转录成RNA的可能性。
2.4 基于生成模型的分子设计
在一个已知药物分子库中训练模型,它将学习生成新的“类药物”分子,用作虚拟筛选中的候选分子。为此而引入的生成模型可能比小分子设计更有影响力。与小分子设计不同的是,对于人类专家来说,预测特定蛋白质突变引发的效应是非常棘手的。使用生成模型可以实现更丰富的蛋白质设计,实现超出当今人类专家能力的方向。
2.5 基于图卷积网络的药物筛选
虚拟筛选可以提供一种经济有效的方法来确定药物发现项目的起点。不需要进行昂贵的实验性的高通量筛选(HTS),而是使用计算方法来评估数百万甚至数千万分子。虚拟筛选方法通常分为基于结构的虚拟筛选和基于配体的虚拟筛选两大类。
基于结构的虚拟筛选:计算方法被用来识别最适合于蛋白质中一个称为结合位点的空腔的分子。分子与蛋白质结合位点的结合常常会抑制蛋白质的功能。例如,被称为酶的蛋白质催化多种生理化学反应。通过识别和优化这些酶作用过程的抑制剂,科学家们已经能够开发出治疗肿瘤、炎症、感染和其他治疗领域的各种疾病的方法。
基于配体的虚拟筛选:搜索那些功能类似于一个或多个已知分子的分子。我们可能希望改善现有分子的功能,避免与已知分子相关的药理学责任,或开发新的知识产权。基于配体的虚拟筛选通常从一系列已知分子开始,这些分子是通过各种实验方法识别的,然后利用计算方法建立基于实验数据的模型,该模型对大量分子进行虚拟筛选,寻找新的化学起点。