细胞通讯分析可以给我们一些细胞类群之间相互调控/交流的信息,这种细胞之间的调控主要是通过受配体结合,传递信号来实现的。不同的分化、疾病过程,可能存在特异的细胞通讯关系,因此阐明这些通讯关系至关重要。
CellPhoneDB配有详实的受配体数据库,其整合了此前的公共数据库,还会手动矫正,以得到更加准确的受配体注释。此外,针对受配体有多个亚基的情况,也进行了注释。下面这张图显示了CellPhoneDB配有的数据库包含多少种分泌蛋白和膜蛋白、蛋白质复合物、受配体关系,以及它们来源于什么数据库。
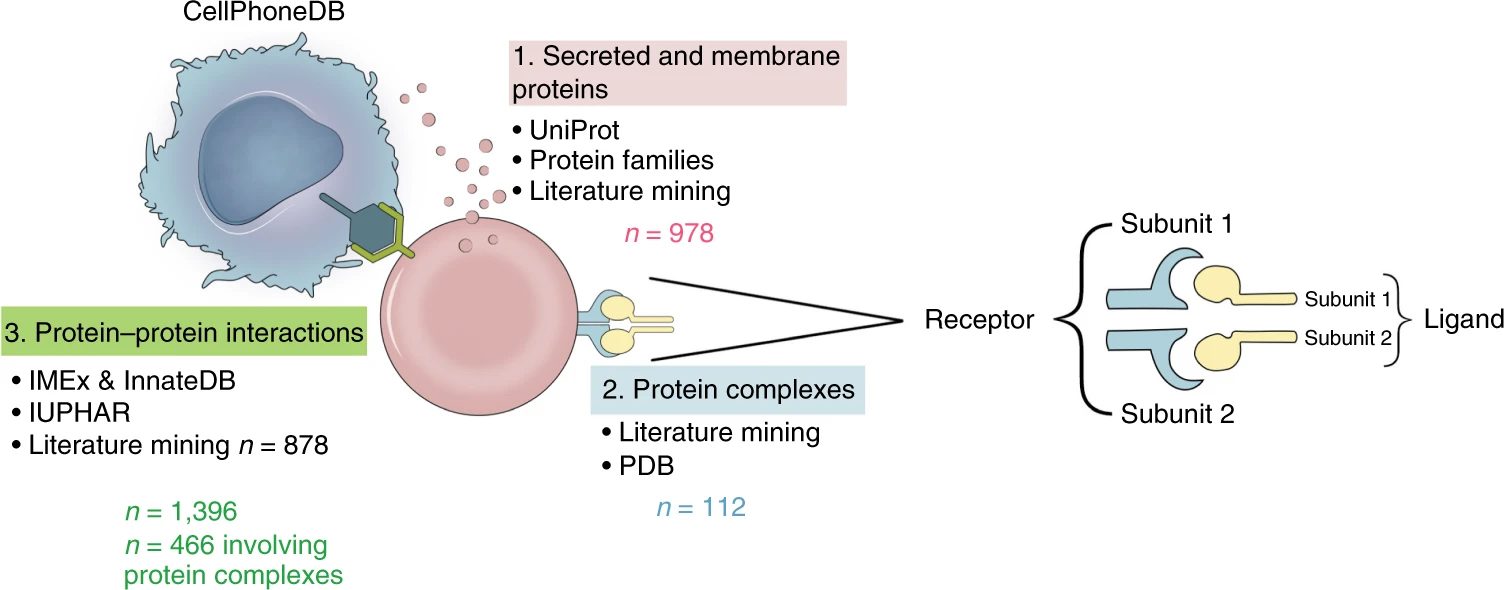
1. CellPhoneDB推断细胞通讯的原理
在给定表达矩阵和细胞注释之后,对于gene1-gene2这个互作关系,计算某一个clusterA里面gene1的表达均值,计算另一个clusterB中gene2的表达均值,二者的均值为MEAN;在随机更换细胞的label之后,依据新的标签,计算“clusterA”里面gene1的表达均值,"clusterB"中gene2的表达均值,再求一个平均值mean,这样的过程重复多次,就可以得到一个mean的分布,即null distribution。MEAN在这个分布中所在的位置以及更极端的位置,构成的占比,就是p值(p值的定义)。所以CellPhoneDB推测两种细胞类型之间显著富集的受配体关系,本质上还是基于一个细胞类型里面的受体表达量,以及另一种细胞类型里面的配体表达量。此外,如果某种关系无处不在(在所有细胞类型之间都很明显),则找不出来。
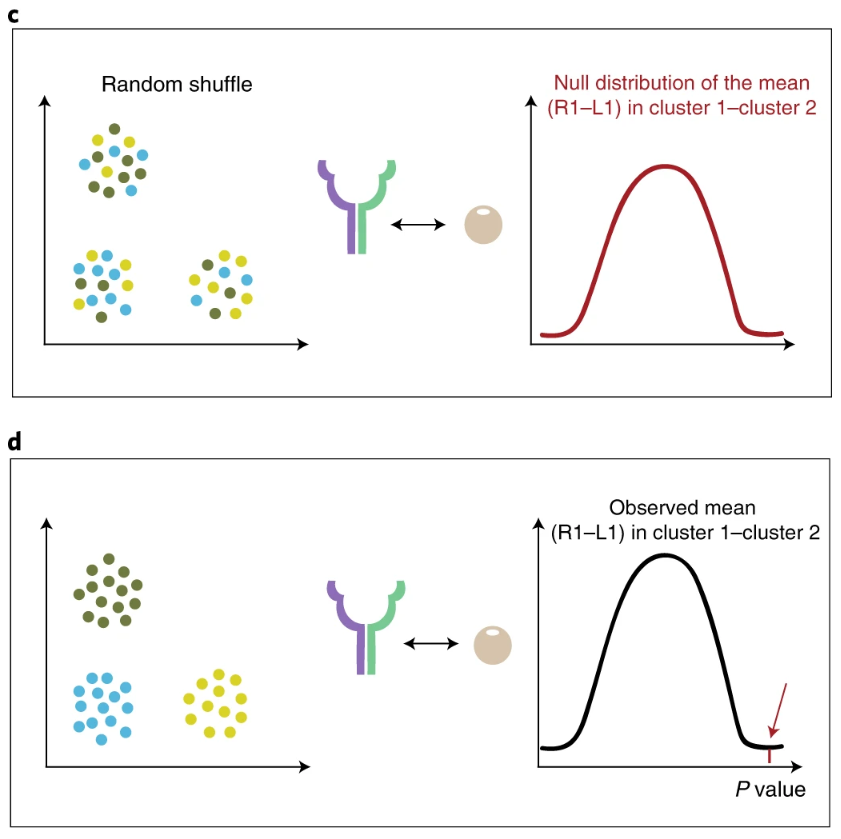
此外还有几个需要注意的地方:
- 大样本时会下采样,只分析1/3的细胞
- 多个亚基时考虑表达低的那一个亚基
- 表达占比达到一定阈值的基因才会被分析,默认是10%
2. 如何展示结果
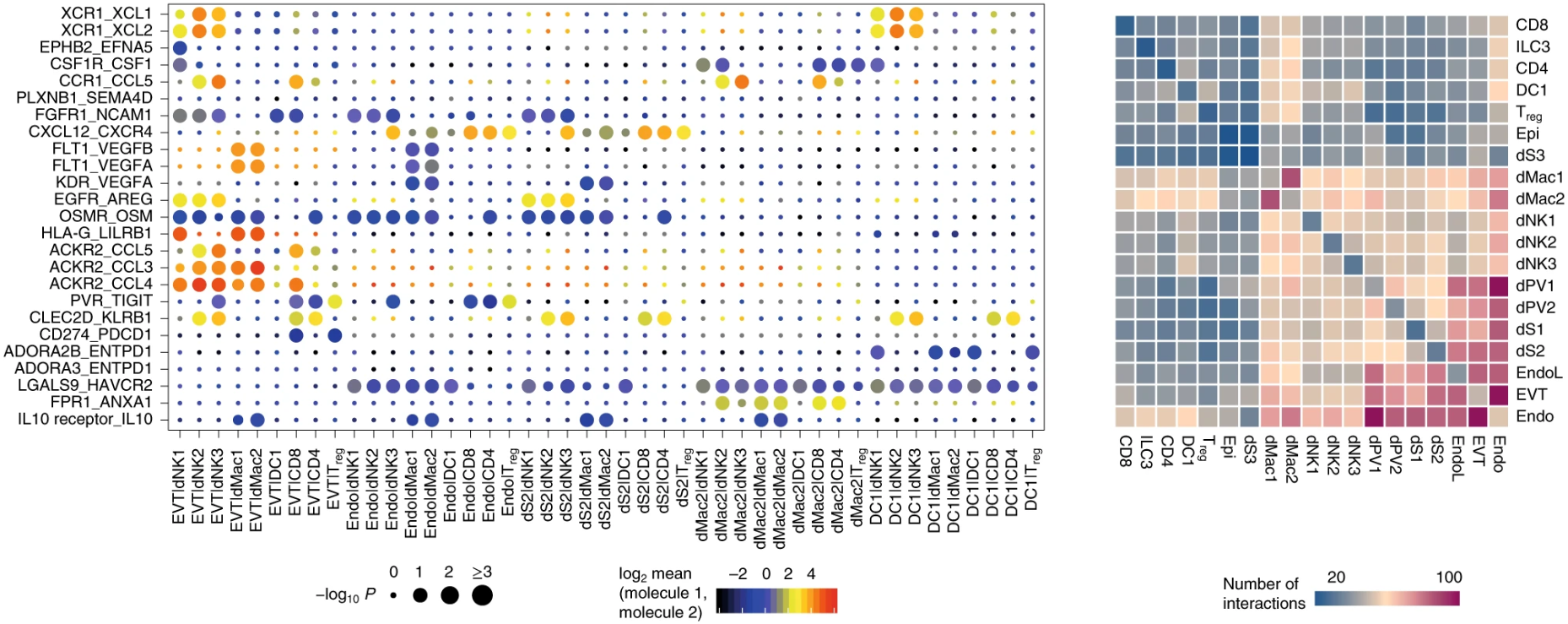
这是原文献给的可视化例子,这里有两个地方需要注意:
- 右边的热图表示细胞类型两两之间的相互作用的数量,我们可以看到沿着对角线,左右是对称的,也就是A-B与B-A的互作数目是一样的,为什么会这样?
- 左边是具体受配体对,细胞对的互作气泡图,点的大小表示显著水平,颜色则是The means of the average expression level of interacting molecule 1 in cluster 1 and interacting molecule 2 in cluster 2 注意到了吗,说的是interacting molecule 1/2,而没有说哪一个是受体哪一个是配体。
原因都和CellPhoneDB内置的gene-gene互作关系列表有关。CellPhoneDB区分不了受体还是配体,对于gene1-gene2,可以是gene1配体gene2受体,也可以是gene1受体gene2配体(如下图)。我个人觉得也是由于这个原因,右边那个热图为了说起来方便,才把不管做受体还是做配体的关系都算作是两种细胞的互作关系,因此A-B和B-A在热图中的数值是一样的(不然横纵坐标写个interacting molecule,看到的人自然会问,这个分子是受体还是配体呢,加一起就省事了——都包含)。
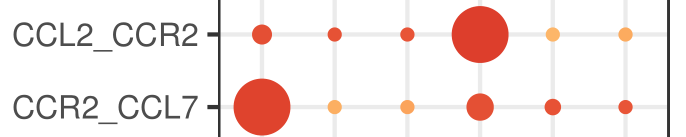
这一点,github有提到:
也是这个原因,我看到文章如果用了CellPhoneDB的话,会留意它的图,如果是用有向图表示细胞群两两之间的关系数量,我会想这样做合不合适(当然是不合适的)
3. 实际分析
公众号后台回复
20210723获取本次演示的测试数据,以及主要的可视化代码。
3.1 输入文件的格式
注释文件
一共两列,Cell列cell_type列,有列名;.csv, .txt后缀都行
表达文件
normalize之后的矩阵,一般简单相除normalize一下就行;.csv, .txt后缀都行
3.2 运行
软件的安装这里就不讲了,创建一个conda环境,pip install下载安装就可以了
运行CellPhoneDB的主代码很简单:
source /home/huangsiyuan/miniconda3/bin/activate cpdb
file_count=/home/huangsiyuan/cpdb/test_normat.txt
file_anno=/home/huangsiyuan/cpdb/test_anno.txt
outdir=/home/huangsiyuan/cpdb/test
if [ ! -d ${outdir} ]; then
mkdir ${outdir}
fi
cellphonedb method statistical_analysis
--counts-data hgnc_symbol
--output-path ${outdir}
--threshold 0.01 #Percentage of cells expressing the specific ligand or receptor
--threads 10
${file_anno} ${file_count}
source /home/huangsiyuan/miniconda3/bin/deactivate cpdb
#如果细胞数太多,可以添加下采样参数,默认只分析1/3的细胞
#--subsampling
#--subsampling-log true #对于没有log转化的数据,还要加这个参数
这一步之后在test文件夹里面会生成4个文件
deconvoluted.txt
means.txt
pvalues.txt
significant_means.txt
其中,
means.txt行是受配体pair,列是细胞pair,值为受体、配体在相应的cluster中表达均值的平均数;pvalues.txt格式与means.txt类似,值为p值;significant_means.txt格式和内容都与means.txt类似,不过仅保留了p值小于0.05的平均数。
4. 结果的可视化
在这一步中,我一般只用到上述的means.txt和pvalues.txt文件
我们还是先仿照文献原文,画出那两张图
library(tidyverse)
library(RColorBrewer)
library(scales)
pvalues=read.table("./test/pvalues.txt",header = T,sep = " ",stringsAsFactors = F)
pvalues=pvalues[,12:dim(pvalues)[2]] #此时不关注前11列
statdf=as.data.frame(colSums(pvalues < 0.05)) #统计在某一种细胞pair的情况之下,显著的受配体pair的数目;阈值可以自己选
colnames(statdf)=c("number")
#排在前面的分子定义为indexa;排在后面的分子定义为indexb
statdf$indexb=str_replace(rownames(statdf),"^.*\.","")
statdf$indexa=str_replace(rownames(statdf),"\..*$","")
#设置合适的细胞类型的顺序
rankname=sort(unique(statdf$indexa))
#转成因子类型,画图时,图形将按照预先设置的顺序排列
statdf$indexa=factor(statdf$indexa,levels = rankname)
statdf$indexb=factor(statdf$indexb,levels = rankname)
statdf%>%ggplot(aes(x=indexa,y=indexb,fill=number))+geom_tile(color="white")+
scale_fill_gradientn(colours = c("#4393C3","#ffdbba","#B2182B"),limits=c(0,20))+
scale_x_discrete("cluster 1 produces molecule 1")+
scale_y_discrete("cluster 2 produces molecule 2")+
theme_minimal()+
theme(
axis.text.x.bottom = element_text(hjust = 1, vjust = NULL, angle = 45),
panel.grid = element_blank()
)
ggsave(filename = "interaction.num.1.pdf",device = "pdf",width = 12,height = 10,units = c("cm"))
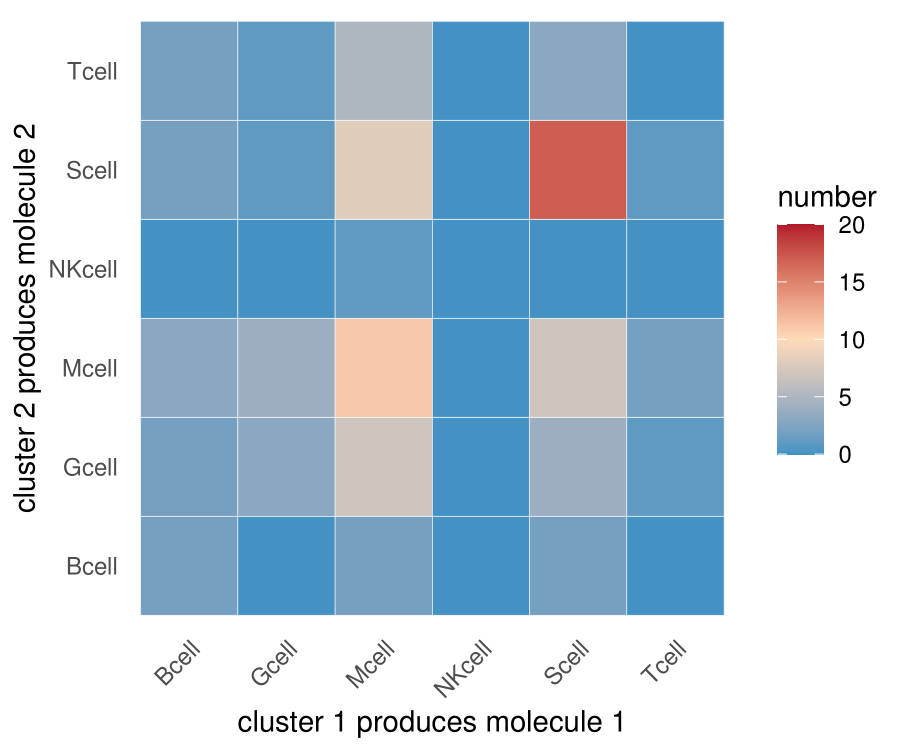
这里与文献中图不一致的地方是,我这个图并不是关于对角线对称的,因为我没有将A-B,B-A的互作关系求和
举个例子
在CellPhoneDB输出的结果中,经统计,A-B有10个显著的互作关系,B-A有20个显著的互作关系【①】。然而A-B的互作其实包含A做配体8次,A做受体2次,B-A的互作其实包含B做配体19次,B做受体1次,所以严格来讲,A和B两种细胞互作,A做配体9次,B做配体21次【②】,这些信息是CellPhoneDB给不了的。当然互作关系还是共计30次【③】。
换言之,文献中对称的图给的信息③,我上面那个图给的信息①,信息②是不知道的(如果肉眼一个一个去看CellPhoneDB数据库中gene1-gene2哪个是受体哪个是配体,还是可以统计出来的)。
因本文篇幅较长,余下的可视化部分将在下一篇展示,敬请期待~
参考文献
[1] Efremova M, Vento-Tormo M, Teichmann S A, et al. CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes[J]. Nature protocols, 2020, 15(4): 1484-1506.
因水平有限,有错误的地方,欢迎批评指正!