在我之前的帖子单细胞分析实录(7): 差异表达分析/细胞类型注释里面,我已经介绍了如何使用SingleR给单细胞数据做注释,当时只讲了SingleR配套的参考集。这次就讲讲如何使用自己定义/找到的基因集做注释,使用场景还是比较多的,比如想根据某篇论文里面的注释结果,给自己的数据做注释。本文配套的视频讲解已上传到B站,新手UP: TOP菌。gongzhong号后台回复20211023可获取本文所用到的示例数据和代码。

本次演示用到的数据集来自2020年发表在Nature Genetics的一篇结直肠癌研究。文中用到了韩国患者(SMC)和比利时患者(KUL3)两套数据集,两套数据平行分析,相互印证。

我以里面的髓系细胞为例,用KUL3数据集中的髓系细胞为参考,来注释SMC里面的髓系细胞
两套数据集中髓系细胞部分的数据,已经跑完Seurat标准流程并整理成rds文件:SMC_mye.rds和KUL3_mye.rds。代码存放在0.R中,这里就不展示了,很简单。代码中fread()用于快速读取4G矩阵文件是可以学习的地方。
mat=fread("GSE132465_GEO_processed_CRC_10X_raw_UMI_count_matrix.txt",select = c("Index",SMC.mye.anno$Index))
之后就是创建参考集,被存储为SummarizedExperiment对象
library(Seurat)
library(tidyverse)
library(SummarizedExperiment)
library(scuttle)
KUL3_mye=readRDS("KUL3_mye.rds")

属性数据框中主要用到cell index和cell annotation这两个信息,此外还需导入表达矩阵
KUL3_mye_count=KUL3_mye[["RNA"]]@counts
pdata=KUL3_mye@meta.data[,c("Index","Cell_subtype")]
rownames(pdata)=pdata$Index
pdata$Index=NULL
colnames(pdata)="ref_label"
KUL3_mye_SE <- SummarizedExperiment(assays=list(counts=KUL3_mye_count),colData = pdata)
#创建SummarizedExperiment对象
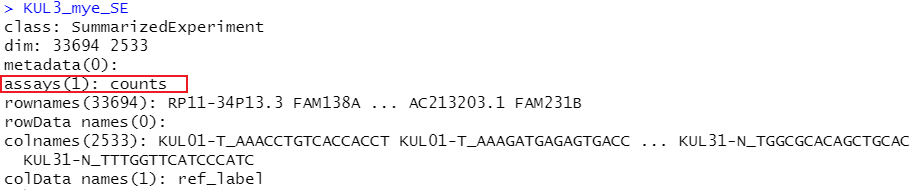
KUL3_mye_SE <- logNormCounts(KUL3_mye_SE)
#Compute log-transformed normalized expression values,要有这一行,对于单细胞数据normalize之后会再算一个log
saveRDS(KUL3_mye_SE,"KUL3_mye_SE.ref.rds")
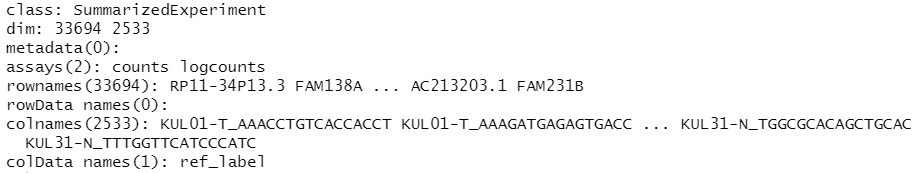
log normalize之后就多了一个logcounts的assay,singleR的官网示例都是基于logcounts这种数据
将这个参考对象保存为rds文件,方便以后多次调用
现在导入我们的待注释数据集和参考数据集
library(tidyverse)
library(Seurat)
library(SingleR)
library(scuttle)
library(SummarizedExperiment)
KUL3_mye_SE=readRDS("KUL3_mye_SE.ref.rds")
SMC_mye=readRDS("SMC_mye.rds")
SMC_mye_count=SMC_mye[["RNA"]]@counts
#需要取不同数据集的基因交集
common_gene <- intersect(rownames(SMC_mye_count), rownames(KUL3_mye_SE))
KUL3_mye_SE <- KUL3_mye_SE[common_gene,]
SMC_mye_count <- SMC_mye_count[common_gene,]
#也要创建SummarizedExperiment对象,以及log normalize
SMC_mye_SE <- SummarizedExperiment(assays=list(counts=SMC_mye_count))
SMC_mye_SE <- logNormCounts(SMC_mye_SE)
#注释代码
singleR_res <- SingleR(test = SMC_mye_SE, ref = KUL3_mye_SE, labels = KUL3_mye_SE$ref_label)
#导出注释结果
anno_df <- as.data.frame(singleR_res$labels)
anno_df$Index <- rownames(singleR_res)
colnames(anno_df)[1] <- 'ref_label_from_KUL3'
#将注释信息添加到Seurat对象
SMC_mye@meta.data=SMC_mye@meta.data%>%inner_join(anno_df,by="Index")
rownames(SMC_mye@meta.data)=SMC_mye@meta.data$Index
DimPlot(SMC_mye, reduction = "tsne", group.by = "Cell_subtype", pt.size=2)+
DimPlot(SMC_mye, reduction = "tsne", group.by = "ref_label_from_KUL3", pt.size=2)
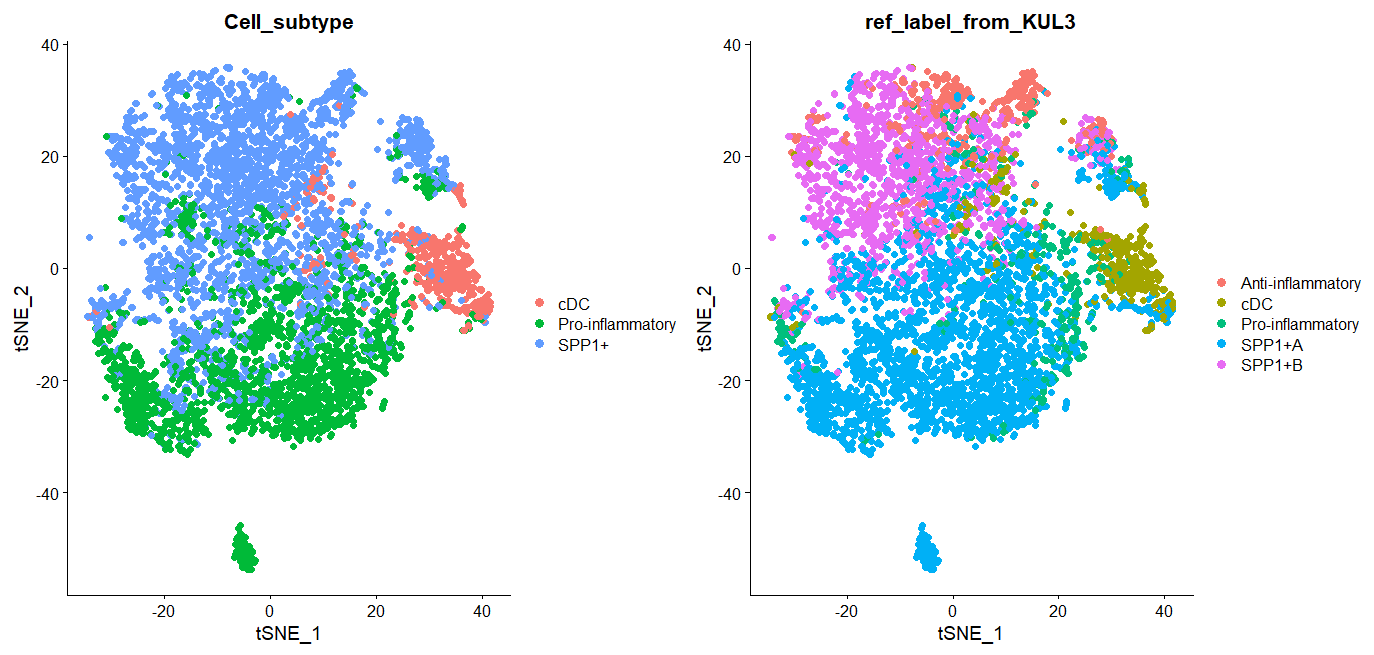
左图是原文给的注释,右图是依据KUL3来注释SMC数据集,可以看出还是有一些不一致的。
像这种依据某个参考集来做注释,对参考集质量要求很高,原文KUL3只有两千髓系细胞,考虑到10X单细胞数据的稀疏性,这个数量是不够的。也可能是软件的限制,在做小类注释时,类与类之间的表达特征其实是比较相似的,软件不一定能精确给出合适的label,相比之下,软件做大类注释一般比较准。
因水平有限,有错误的地方,欢迎批评指正!