之前写过三篇和CNV相关的帖子,如果你做肿瘤单细胞转录组,大概率看过:
单细胞分析实录(11): inferCNV的基本用法
单细胞分析实录(12): 如何推断肿瘤细胞
单细胞分析实录(13): inferCNV结合UPhyloplot2分析肿瘤进化
其中,第三篇帖子里面有两个注释代码,可以在基因和染色体长短臂两个层面对CNV做注释
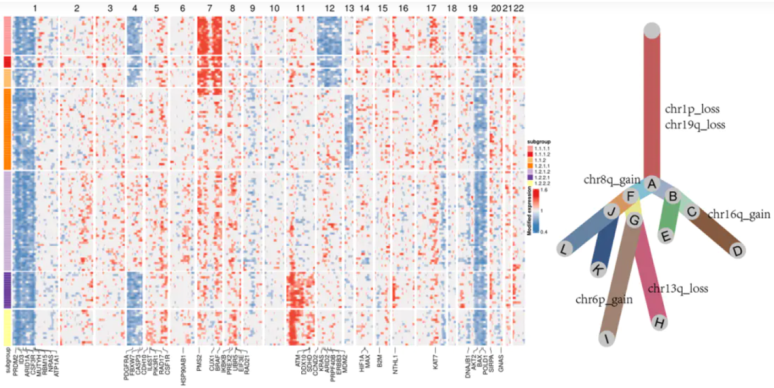
这次对tree_anno.R代码做了更新,简单来说就是对原始CNV region的长度做了限制,最后的结果会输出更少更明显的CNV

之前在我这里拿过代码的读者如果需要,可以直接找我拿更新后的代码(tree_anno.new.R),以及下面的代码(cnv_heatmap.R)。
另外,需要说明的是,距离我上次发布inferCNV帖子(2021年4月)已经半年了,前些天有读者跟我说,新版的inferCNV(读者给我看的是inferCNV v1.8.1),在给肿瘤细胞分组时,默认的方法已经变了,cell_groupings文件中已经没有tree相关的信息了。如果仍然想得到肿瘤进化树,需要改
tumor_subcluster_partition_method参数为random_trees,默认是leiden,后者速度快但没有tree信息。
以下是这篇帖子的主要内容。
参考的还是之前提到的两篇NC文章:

ref1: Single-cell RNA landscape of intratumoral heterogeneity and immunosuppressive microenvironment in advanced osteosarcoma
ref2: Single-cell analysis reveals new evolutionary complexity in uveal melanoma
inferCNV的热图可以反映单个样本的CNV情况,这个热图可以反映多个样本的CNV情况,而且简化到染色体长短臂。在样本比较多时,还是比较有用的。
这次的代码仍然依赖inferCNV的输出信息,仍然是"subclusters"模式。(inferCNV相关的用法参考我之前的帖子,这里不多说)
最终的效果图:

图中列是样本,行是CNV type (chr1p_loss这种)。每一个方块代表,该样本具有这种CNV的细胞在所有肿瘤细胞中的占比。
因水平有限,有错误的地方,欢迎批评指正!