转自:
https://blog.csdn.net/herokoking/article/details/79276939
Pileup 格式最初是由Sanger Institute的Tony Cox 和 Zemin Ning 使用的,描述了染色体上每个位置的碱基信息。 可以用来 SNP/indel calling, 也可以直接用眼睛看一下排列的情况。
Pileup 文件一般是由SAMtools从sorted bam 文件生成。
samtools mpileup -f ../crrbwaidx/crr5ref.fa -Q 15 -q 20 4_S4_L001.sorted.bam -o 4_S4_L001.pileup
mpileup 就是SAMtools生成pileup文件的命令(SAMtools 文档):
-f 后面是参考序列或者基因组
-q 是mapping quality的最小值, 默认值是0
-Q 是base quality的最小值,默认值是13
然后是输入文件*.sorted.bam, -o 后面是输出文件*.pileup
pileup 格式有几个变体,下面的是由SAMtools输出的默认格式:
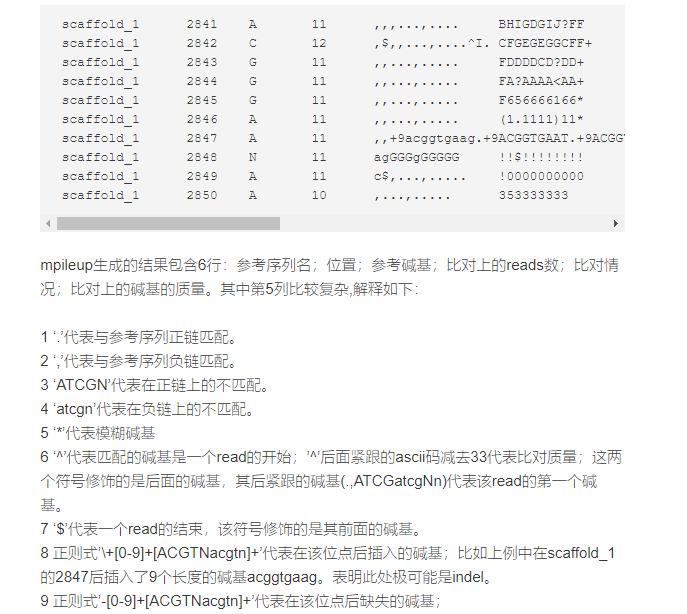
seq1 272 T 24 ,.$.....,,.,.,...,,,.,..^+. <<<+;<<<<<<<<<<<=<;<;7<&seq1 273 T 23 ,.....,,.,.,...,,,.,..A <<<;<<<<<<<<<3<=<<<;<<+seq1 274 T 23 ,.$....,,.,.,...,,,.,... 7<7;<;<<<<<<<<<=<;<;<<6seq1 275 A 23 ,$....,,.,.,...,,,.,...^l. <+;9*<<<<<<<<<=<<:;<<<<seq1 276 G 22 ...T,,.,.,...,,,.,.... 33;+<<7=7<<7<&<<1;<<6<seq1 277 T 22 ....,,.,.,.C.,,,.,..G. +7<;<<<<<<<&<=<<:;<<&<seq1 278 G 23 ....,,.,.,...,,,.,....^k. %38*<<;<7<<7<=<<<;<<<<<seq1 279 C 23 A..T,,.,.,...,,,.,..... ;75&<<<<<<<<<=<<<9<<:<<每一行由染色体,参考碱基在染色体中的位置(reference position),参考碱基(reference base),覆盖该位点的reads的个数,覆盖该位点的碱基(read bases) 和碱基质量(base qualities),之间由tab分开。
在read base那一列,每个点(dot)代表这个碱基正向匹配到reference base, 每个逗号代表该碱基反向匹配到reference base. 如果是‘ACGTN’,说明这个碱基是个正向的mismatch,如果是‘actgn', 说明这个碱基是个反向的mismatch。这个样式 '+ or - [0-9]+[ACGTNacgtn]+'是在这个reference position和下一个reference position之间的插入(insertion)或缺失(deletion)突变。加号代表插入,减号代表缺失,数字是插入或缺失的长度,数字后面的’ACGTN or acgtn'就是插入或缺失的碱基。下面第一行是一个插入的例子,第二行是两个缺失的例子(有五个reads都出现了两个的缺失):
chr1 12994 G 20 ......,,,+1c,,,,,,,,,,, KK<KFKFAKKKFKKKKK7KKchr1 13656 C 18 .-2AG....,-2ag,,-2ag.,.,-2ag....,-2ag, FKKKKKKKKF7KKKKKK<在read base那一列,‘^'是每条reads的开始,’^'后面跟的符号" ' "的ASCII十进制值减去33就是这条reads的mapping quality. '$'是每条reads的结束。如下:
chr1 13005 A 21 .....,,,,,,,,,,,,,^',^',^', 7KFFKFF<KKKKKK7K<KEEEchr1 13006 G 24 .$.....,,,,,,,,,,,,,,,,,^'. >KKFAKK7FKKKKKKKKFKKKFKB
reads开始和结束的标志是受到了Phil Green's CALF format 的启发。通过开始和结束的标志可以从pileup文件重建reads.
SAMtools 可以选择性的把mapping quality加到输出结果的每一行,这样会使输出的文件更大,但当只有一部分位点被提取出来时,这样做就有必要了(因为只提取一部分位点,开始的标志'^'就可能不会被包含着提取的位点,就没法知道每个reads base的mapping quality了.)。
以下来自:http://www.chenlianfu.com/?p=1399