基本概念
Biostrings包很重要的3个功能是进行Pairwise sequence alignment 和Multiple sequence alignment及 Pattern finding in a sequence
序列比对一般有2个过程:
1)构建计分矩阵公式(the scoring matrix formulation)
2)比对(alignment itself)
global alignment methods (全局比对):align every residue in the sequences ,例如Needleman-Wunsch algorithm.
local alignment technique(局部比对): align regions of high similarity in the sequences,例如Smith-Waterman algorithm
安装
if("Biostrings" %in% rownames(installed.packages()) == FALSE) {source("http://bioconductor.org/biocLite.R");biocLite("Biostrings")}
suppressMessages(library(Biostrings))
ls('package:Biostrings')
----------------Pairwise sequence alignment---------------
步骤:首先构建罚分规则,然后按照规则进行比对。用pairwiseAlignment()函数
举例1:核酸序列
(myScoringMat <- nucleotideSubstitutionMatrix(match = 1, mismatch = -1, baseOnly = TRUE))#构建罚分规则
gapOpen <- 2 #gap分为2
gapExtend <- 1 #延伸gap分为1
sequence1 <- "GAATTCGGCTA" #序列1
sequence2 <- "GATTACCTA" #序列2
myAlignment <- pairwiseAlignment(sequence1, sequence2,
substitutionMatrix = myScoringMat, gapOpening = gapOpen,
gapExtension = gapExtend, type="global", scoreOnly = FALSE) #进行比对
myAlignment

举例2:对蛋白序列进行比对
蛋白比对会更复杂,因此模型更多,
data(package="Biostrings") #查看所有数据集 data(BLOSUM62) #这里选择BLOSUM62数据 subMat <- "BLOSUM62" #赋值 gapOpen <- 2 gapExtend <- 1 sequence1 <- "PAWHEAE" sequence2 <- "HEAGAWGHE"
myAlignProt <- pairwiseAlignment(sequence1, sequence2, substitutionMatrix = subMat, gapOpening = gapOpen, gapExtension = gapExtend, type="global", scoreOnly = FALSE) #全局比对
myAlignProt2 <- pairwiseAlignment(sequence1, sequence2, substitutionMatrix = subMat, gapOpening = gapOpen, gapExtension = gapExtend, type="local",scoreOnly = FALSE) ##局部比对
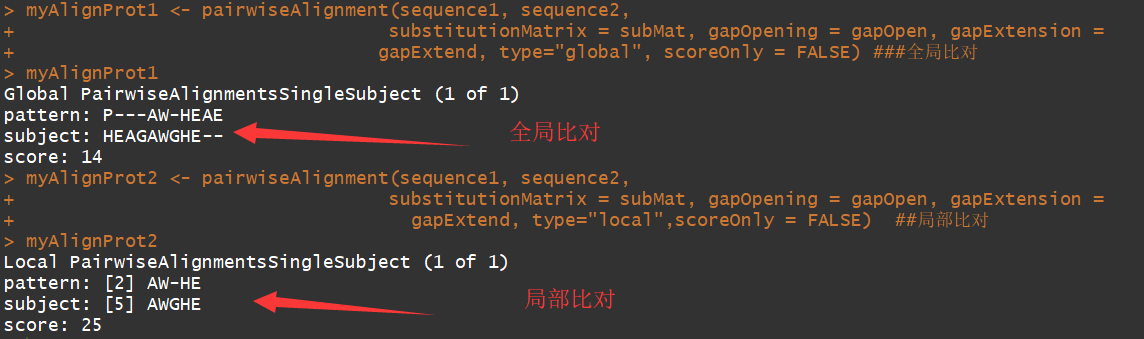
可以看到局部比对返回的是,高度相似的序列部分.
3)可视化,对于序列可以用最经典的对角线来可视化(以人和黑猩猩的hemoglobin beta为例)
library(seqinr) # 为了读取fasta序列
myseq <- read.fasta(file = "F:/R/Bioconductor/biostrings/prtein_example_seq.fas")
dotPlot(myseq[[1]], myseq[[2]], col=c("white", "red"), xlab="Human", ylab="Chimpanzee")

##########Multiple sequence alignment############
一般多序列比对可以用于进化分析
install.packages("muscle") #需要安装该包,因为该包在我的版本上没法安装,所以这里就不讲了
library(muscle)
######Phylogenetic analysis and tree plotting########
这里先不做分析
#########blast格式的解析######
install.packages("RFLPtools",dependencies=TRUE)
library(RFLPtools)
data(BLASTdata) #先查看数据集了解一下相关数据格式情况
head(BLASTdata)
colnames(BLASTdata)
DIR <- system.file("extdata", package = "RFLPtools") #用自带数据集
MyFile <- file.path(DIR, "BLASTexample.txt")
MyBLAST <- read.blast(file = MyFile)
mySimMat <- simMatrix(MyBLAST) #可以根据blast结果用来生成相似性矩阵,太厉害了
#########Pattern finding in a sequence######
library(Biostrings)
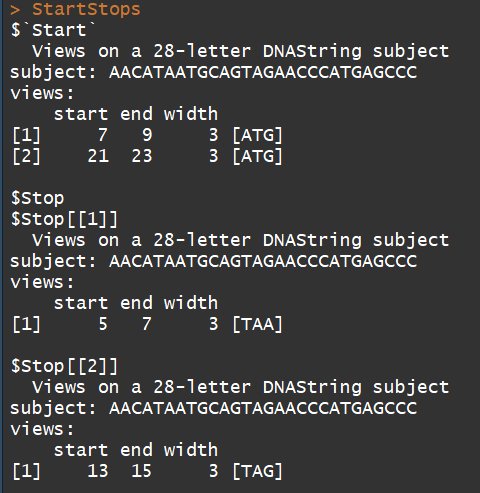
mynucleotide <- DNAString("aacataatgcagtagaacccatgagccc")
matchPattern(DNAString("ATG"), mynucleotide) #示例1
matchPattern("TAA", mynucleotide) #示例2
##以下函数可以用来寻找orf(需要修改)
myCodonFinder <- function(sequence){
startCodon = DNAString("ATG") # 指定起始密码子
stopCodons = list("TAA", "TAG", "TGA") # 指定终止密码子
codonPosition = list() #initialize the output to be returned as a list
codonPosition$Start = matchPattern(startCodon, sequence) # search start codons
x=list()
for(i in 1:3){ # iterate over all stop codons
x[[i]]= matchPattern(DNAString(stopCodons[[i]]), sequence)
codonPosition$Stop=x
}
return(codonPosition) # returns results
}
StartStops <- myCodonFinder(mynucleotide)