微生物群落多样性测序与功能分析
1.几个概念:16S rDNA(或16S rRNA);OTU;测序区段。
1.3测序区段:由于16S rDNA较长(1.5kb),我们只能对其中经常变化的区域也就是可变区进行测序。16S rDNA包含有9个可变区,分别是v1-v9。一般我们对v3-v4双可变区域进行扩增和测序,也有对v1-v3区进行扩增测序。
2.工具/原料:
2.1样本:16S rDNA测序首先需要提取环境样品的DNA,这些DNA可以来自土壤、粪便、空气或水体等任何来源。
2.2提取DNA:提取DNA后需要经过质检和纯化,一般16S rDNA测序扩增对DNA的总量要求并不高,总量大于100ng,浓度大于10ng/ul一般都可以满足要求。如果是来自和寄主共生的环境如昆虫的肠道微生物,提取时可能包括了寄主本身的大量DNA,对DNA的总量要求会提高。微生物菌群多样性测序受DNA提取和扩增影响很大,不同的扩增区段和扩增引物甚至PCR循环数的差异都会对结果有所影响。因而建议同一项目不同样品的都采用相同的条件和测序方法,这样相互之间才存在可比性。
2.3测序:完成PCR之后的产物一般可以直接上测序仪测序,在上机测序前我们需要对所有样本进行定量和均一化,通常要进行荧光定量PCR。完成定量的样品混合后就可以上机测序。(16S rDNA测序目前可以采用多种不同的测序仪进行测序,包括罗氏的454,Illumina的Novoseq, MiSeq,Hiseq,Life的 PGM 或 Pacbio 以及 nanopore 的三代测序仪。不同的仪器各有优缺点,目前最主流的是Illumina公司的MiSeq,因为其在通量、长度和价格三者之间最为平衡。MiSeq 测序仪可以产生 2x300 bp 的测序读长, Hiseq 和 Novoseq 可以生成 2x250bp 或者 2x150bp 的测序读长,且通量较大。)
3.方法:
16S rDNA分析基本流程:数据预处理;OTU分析;样本差异分析
3.1原始数据处理:
原始测序数据需要去除接头序列,根据 overlap 软件并将双端测序序列进行拼接成单条序列,同时对序列质量进行质控和过滤。提供已知数据库 GreenGenes 作为参考,去除嵌合体序列得到最终可用的序列。
提取出的数据以 fastq 格式保存,每个样本有 fq1 和 fq2两个文件,里面为测序两端的 reads,序列按顺序一一对应。
原始fastq格式是一个文本格式用于存储生物序列(通常是核酸序列)和其测序对应的质量值。这些序列以及质量信息用ASCII字符标识。
3.2OTU分类和统计:
OTU(operational taxonomic units) 是在系统发生学研究或群体遗传学研究中,为了便于进行分析,人为给某一个分类单元(品系,种,属,分组等)设置的同一标志。通常按照 97% 的相似性阈值将序列划分为不同的 OTU,每一个 OTU 通常被视为一个微生物物种。相似性小于97%就可以认为属于不同的种,相似性小于93%-95%,可以认为属于不同的属。样品中的微生物多样性和不同微生物的丰度都是基于对OTU的分析。
使用QIIME(version 1.8.0)工具包进行统计注释。
使用QIIME(version 1.9.0, http://bio.cug.edu.cn/qiime/)的ucluster方法根据97%的序列相似度将所有序列进行同源比对并聚类成operational taxonomic units (OTUs)。然后与数据库GreenGenes(version gg_13_8, http://greengenes.lbl.gov/cgi-bin/JD_Tutorial/nph-16S.cgi)进行比对,比对方法uclust,identity 0.9 。
然后对每个OTUs进行reads数目统计。
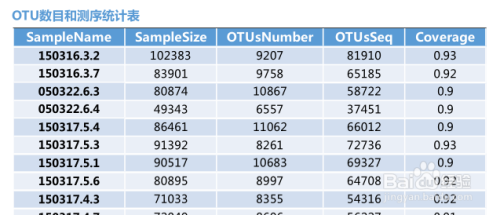
下面的2个表,其中一个表是对每个样本的测序数量和OTU数目进行统计,并且在表栺中列出了测序覆盖的完整度(显示前10个样本)。
另一个表是对每个样本在分类字水平上的数量进行统计,并且在表栺中列出了在每个分类字水平上的物种数目(显示前10个样本)。
可以看到绝大部分的OTU都分类到了属(Genus),也有很多分类到了种(Species)。但是仍然有很多无法完全分类到种一级,这是由于环境微生物本身存在非常丰富的多样性,还有大量的菌仍然没有被测序和发现。
测序数目统计表主要是对每个样本的测序数量和OTU数目进行统计,并且在表格中列出了测序覆盖的完整度(显示前10个样本,如果样本超过10个,请查看结果中otu_stat.txt文件)
其中 SampleName表示样本名称;SampleSize表示样本序列总数;OTUsNumber表示注释上的OTU数目;OTUsSeq表示注释上OTU的样本序列总数。
Coverage是指各样品文库的覆盖率,其数值越高,则样本中序列没有被测出的概率越低。该指数实际反映了本次测序结果是否代表样本的真实情况。
计算公式为:C=1-n1/N 其中n1 = 只含有一条序列的OTU的数目; N = 抽样中出现的总的序列数目。
分类水平统计表主要是对每个样本在分类学水平上的数量进行统计,并且在表格中列出了在每个分类学水平上的物种数目(只显示前10个样本,如果样本超过10个,请查看结果中taxon_all.txt文件)
其中SampleName表示样本名称;Phylum表示分类到门的OTU数量;Class表示分类到纲的OTU数量;Order表示分类到目的OTU数量;Family表示分类到科的OTU数量;Genus表示分类到属的OTU数量;Species表示分类到种的OTU数量。