原文链接:http://tecdat.cn/?p=23378
原文出处:拓端数据部落公众号
1 介绍
在本文中,我们将研究以下主题
- 证明为什么低维预测模型在高维中会失败。
- 进行主成分回归(PCR)。
- 使用glmnet()进行岭回归、lasso 和弹性网elastic net
- 对这些预测模型进行评估
1.1 数据集
在本文中,我们将使用基因表达数据。这个数据集包含120个样本的200个基因的基因表达数据。这些数据来源于哺乳动物眼组织样本的微阵列实验。
该数据集由两个对象组成:
genes: 一个120×200的矩阵,包含120个样本(行)的200个基因的表达水平(列)。trim32: 一个含有120个TRIM32基因表达水平的向量。
-
##查看刚刚加载的对象
-
str(genes)

![]()
这个练习的目的是根据微阵列实验中测量的200个基因的表达水平预测TRIM32的表达水平。为此,需要从构建中心化数据开始。我们将其存储在两个矩阵X和Y中。
-
X <- scale(gen, center = TRUE, scale = TRUE)
-
Y <- scale(tri, center = TRUE)
请记住,标准化可以避免量纲上的差异,使一个变量(基因)在结果中具有更大的影响力。对于Y向量,这不是一个问题,因为我们讨论的是一个单一的变量。不进行标准化会使预测结果可解释为 "偏离平均值"。
1.2 奇异性诅咒
我们首先假设预测因子和结果已经中心化,因此截距为0。我们会看到通常的回归模型。
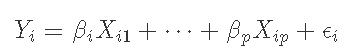
我们的目标是得到β的最小二乘估计值,由以下公式给出
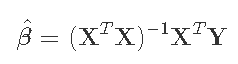
其中p×p矩阵(XTX)-1是关键! 为了能够计算出XTX的逆,它必须是满秩p。我们检查一下。
-
dim(X) # 120 x 200, p > n!
-
#> [1] 120 200
-
qr(X)$rank
-
#> [1] 119
-
-
XtX <- crossprod(X) # 更有效地计算t(X) %*% X
-
qr(XtX)$rank
-
#> [1] 119
-
-
# 尝试用solve进行求解。
-
solve(XtX)

我们意识到无法计算(XTX)-1,因为(XTX)的秩小于p,因此我们无法通过最小二乘法得到β^! 这通常被称为奇异性问题。
2 主成分回归
处理这种奇异性的第一个方法是使用主成分绕过它。由于min(n,p)=n=120,PCA将得到120个成分,每个成分是p=200个变量的线性组合。这120个PC包含了原始数据中的所有信息。我们也可以使用X的近似值,即只使用几个(k<120)PC。因此,我们使用PCA作为减少维度的方法,同时尽可能多地保留观测值之间的变化。一旦我们有了这些PC,我们就可以把它们作为线性回归模型的变量。
2.1对主成分PC的经典线性回归
我们首先用prcomp计算数据的PCA。我们将使用一个任意的k=4个PC的截止点来说明对PC进行回归的过程。
-
k <- 4 #任意选择k=4
-
Vk <- pca$rotation[, 1:k] # 载荷矩阵
-
Zk <- pca$x[, 1:k] # 分数矩阵
-
-
# 在经典的线性回归中使用这些分数
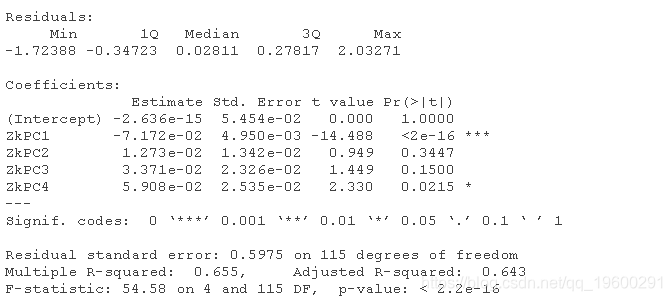
由于X和Y是中心化的,截距近似为0。
输出结果显示,PC1和PC4的β估计值与0相差很大(在p<0.05),但是结果不能轻易解释,因为我们没有对PC的直接解释。
2.2 使用软件包
PCR也可以直接在数据上进行(所以不必先手动进行PCA)。在使用这个函数时,你必须牢记几件事。
- 要使用的成分(PC)的数量是通过参数ncomp来确定
- 该函数允许你首先对预测因子进行标准化(set scale = TRUE)和中心化(set center = TRUE)(在这里的例子中,XX已经被中心化和标准化了)。
你可以用与使用lm()相同的方式使用pcr()函数。使用函数summary()可以很容易地检查得出的拟合结果,但输出结果看起来与你从lm得到的结果完全不同。
#X已经被标准化和中心化了

首先,输出显示了数据维度和使用的拟合方法。在本例中,是基于SVD的主成分PC计算。summary()函数还提供了使用不同数量的成分在预测因子和响应中解释方差的百分比。例如,第一个PC只解释了所有方差的61.22%,或预测因子中的信息,它解释了结果中方差的62.9%。请注意,对于这两种方法,主成分数量的选择都是任意选择的,即4个。
在后面的阶段,我们将研究如何选择预测误差最小的成分数。
3 岭回归、Lasso 和弹性网Elastic Nets
岭回归、Lasso 回归和弹性网Elastic Nets都是密切相关的技术,基于同样的想法:在估计函数中加入一个惩罚项,使(XTX)再次成为满秩,并且是可逆的。可以使用两种不同的惩罚项或正则化方法。
- L1正则化:这种正则化在估计方程中加入一个γ1‖β‖1。该项将增加一个基于系数大小绝对值的惩罚。这被Lasso回归所使用。
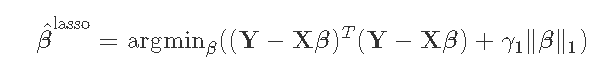
- L2正则化:这种正则化在估计方程中增加了一个项γ2‖β‖22。这个惩罚项是基于系数大小的平方。这被岭回归所使用。
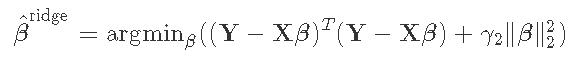
弹性网结合了两种类型的正则化。它是通过引入一个α混合参数来实现的,该参数本质上是将L1和L2规范结合在一个加权平均中。
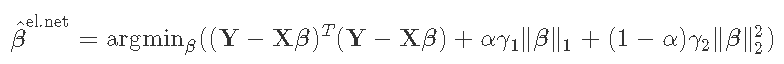
4 练习:岭回归的验证
在最小平方回归中,估计函数的最小化![]() 可以得到解
可以得到解![]() 。
。
对于岭回归所使用的惩罚性最小二乘法准则,你要最小化![]() ,可以得到解
,可以得到解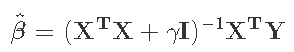 。
。
其中II是p×p的识别矩阵。
脊参数γ将系数缩减为0,γ=0相当于OLS(无缩减),γ=+∞相当于将所有β^设置为0。最佳参数位于两者之间,需要由用户进行调整。
习题
使用R解决以下练习。
1. 验证![]() 秩为200,对于任何一个
秩为200,对于任何一个 ![]() .
.
-
-
gamma <- 2 #
-
-
# 计算惩罚矩阵
-
XtX_gammaI <- XtX + (gamma * diag(p))
-
dim(XtX_gammaI)
-
#> [1] 200 200
-
qr(XtX_gammaI)$rank == 200 #
-
#> [1] TRUE
2. 检查![]() 的逆值是否可以计算出来。
的逆值是否可以计算出来。
-
# 是的,可以被计算。
-
XtX_gammaI_inv <- solve(XtX_gammaI)
3. 最后,计算![]() 。
。
-
## 计算岭β估计值
-
## 使用`drop`来删除维度并创建向量
-
length(ridge_betas) # 每个基因都有一个
-
#> [1] 200

我们现在已经手动计算了岭回归的估计值。
5 用glmnet进行岭回归和套索lasso回归
glmnet允许你拟合所有三种类型的回归。使用哪种类型,可以通过指定alpha参数来决定。对于岭回归,你将alpha设置为0,而对于套索lasso回归,你将alpha设置为1。其他介于0和1之间的α值将适合一种弹性网的形式。这个函数的语法与其他的模型拟合函数略有不同。你必须传递一个x矩阵以及一个y向量。
控制惩罚 "强度 "的gamma值可以通过参数lambda传递。函数glmnet()还可以进行搜索,来找到最佳的拟合伽马值。这可以通过向参数lambda传递多个值来实现。如果不提供,glmnet将根据数据自己生成一个数值范围,而数值的数量可以用nlambda参数控制。这通常是使用glmnet的推荐方式,详见glmnet。
示范:岭回归
让我们进行岭回归,以便用200个基因探针数据预测TRIM32基因的表达水平。我们可以从使用γ值为2开始。
-
glmnet(X, Y, alpha = 0, lambda = gamma)
-
-
#看一下前10个系数

第一个系数是截距,基本上也是0。但γ的值为2可能不是最好的选择,所以让我们看看系数在γ的不同值下如何变化。
我们创建一个γ值的网格,也就是作为glmnet函数的输入值的范围。请注意,这个函数的lambda参数可以采用一个值的向量作为输入,允许用相同的输入数据但不同的超参数来拟合多个模型。
-
grid <- seq(1, 1000, by = 10) # 1到1000,步骤为10
-
# 绘制系数与对数 lambda序列的对比图!
-
plot(ridge_mod_grid)
-
# 在gamma = 2处添加一条垂直线
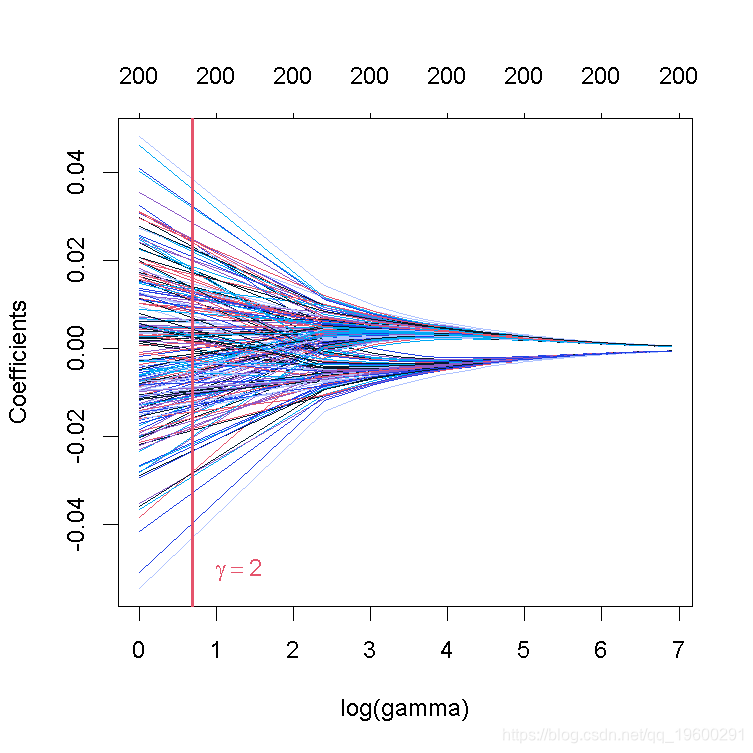
这张图被称为系数曲线图,每条彩线代表回归模型中的一个系数β^,并显示它们如何随着γ(对数)1值的增加而变化。
请注意,对于更高的γ值,系数估计值变得更接近于0,显示了岭惩罚的收缩效应。
与PC回归的例子类似,我们相当随意地选择了γ=2和网格。我们随后会看到,如何选择γ,使预测误差最小。
6 练习: Lasso 回归
Lasso 回归也是惩罚性回归的一种形式,但我们没有像最小二乘法和岭回归那样的β^的分析解。为了拟合一个Lasso 模型,我们再次使用glmnet()函数。然而,这一次我们使用的参数是α=1
任务
1. 验证设置α=1确实对应于使用第3节的方程进行套索回归。
2. 用glmnet函数进行Lasso 套索回归,Y为因变量,X为预测因子。
你不必在这里提供一个自定义的γ(lambda)值序列,而是可以依靠glmnet的默认行为,即根据数据选择γ值的网格。
-
# 请注意,glmnet()函数可以自动提供伽马值
-
# 默认情况下,它使用100个lambda值的序列
3. 绘制系数曲线图并进行解释。
plot(lasso_model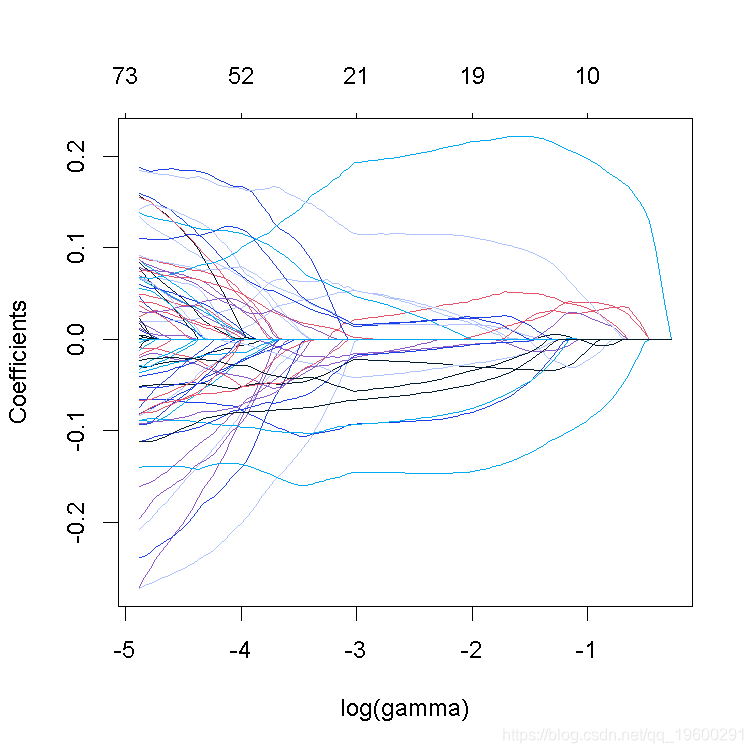
请注意,非零系数的数量显示在图的顶部。在lasso回归的情况下,与岭回归相比,正则化要不那么平滑,一些系数在较高的γ值下会增加,然后急剧下降到0。与岭回归相反,lasso最终将所有系数缩减为0。
7 预测模型的评估和超参数的调整
首先,我们将把我们的原始数据分成训练集和测试集来验证我们的模型。训练集将被用来训练模型和调整超参数,而测试集将被用来评估我们最终模型的样本外性能。如果我们使用相同的数据来拟合和测试模型,我们会得到有偏见的结果。
在开始之前,我们使用set.seed()函数来为R的随机数生成器设置一个种子,这样我们就能得到与下面所示完全相同的结果。一般来说,在进行交叉验证等包含随机性元素的分析时,设置一个随机种子是很好的做法,这样所得到的结果就可以在以后的时间里重现。
我们首先使用sample()函数将样本集分成两个子集,从原来的120个观测值中随机选择80个观测值的子集。我们把这些观测值称为训练集。其余的观察值将被用作测试集。
-
set.seed(1)
-
# 从X的行中随机抽取80个ID(共120个)。
-
trainID <- sample(nrow(X), 80)
-
-
# 训练数据
-
trainX <- X[trainID, ]
-
trainY <- Y[trainID]
-
-
# 测试数据
-
testX <- X[-trainID, ]
-
testY <- Y[-trainID]
为了使以后的模型拟合更容易一些,我们还将创建2个数据框,将训练和测试数据的因变量和预测因素结合起来。

7.1 模型评估
我们对我们的模型的样本外误差感兴趣,即我们的模型在未见过的数据上的表现如何。 这将使我们能够比较不同类别的模型。对于连续结果,我们将使用平均平方误差(MSE)(或其平方根版本,RMSE)。
该评估使我们能够在数据上比较不同类型模型的性能,例如PC主成分回归、岭回归和套索lasso回归。然而,我们仍然需要通过选择最佳的超参数(PC回归的PC数和lasso和山脊的γ数)来找到这些类别中的最佳模型。为此,我们将在训练集上使用k-fold交叉验证。
7.2 调整超参数
测试集只用于评估最终模型。为了实现这个最终模型,我们需要找到最佳的超参数,即对未见过的数据最能概括模型的超参数。我们可以通过在训练数据上使用k倍交叉验证(CVk)来估计这一点。
对于任何广义线性模型,CVk估计值都可以用cv.glm()函数自动计算出来。
8 例子: PC回归的评估
我们从PC回归开始,使用k-fold交叉验证寻找使MSE最小的最佳PC数。然后,我们使用这个最优的PC数来训练最终模型,并在测试数据上对其进行评估。
8.1 用k-fold交叉验证来调整主成分的数量
方便的是,pcr函数有一个k-fold交叉验证的实现。我们只需要设置validation = CV和segments = 20就可以用PC回归进行20折交叉验证。如果我们不指定ncomp,pcr将选择可用于CV的最大数量的PC。
请注意,我们的训练数据trainX由80个观测值(行)组成。如果我们执行20折的CV,这意味着我们将把数据分成20组,所以每组由4个观测值组成。在每个CV周期中,有一个组将被排除,模型将在剩余的组上进行训练。这使得我们在每个CV周期有76个训练观测值,所以可以用于线性回归的最大成分数是75。
-
## 为可重复性设置种子,kCV是一个随机的过程!
-
set.seed(123)
-
-
##Y ~ . "符号的意思是:用数据中的每个其他变量来拟合Y。
-
summary(pcr_cv)
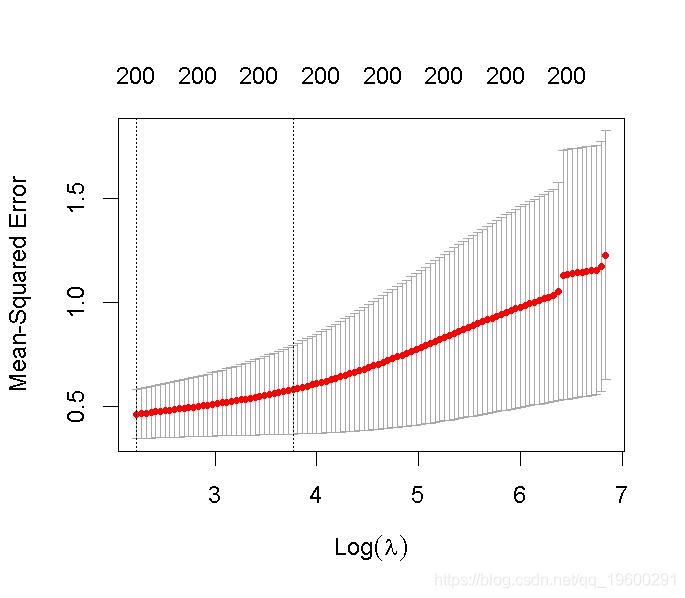
我们可以绘制每个成分数量的预测均方根误差(RMSEP),如下所示。
plot(pcr_cv, plottype = "validation")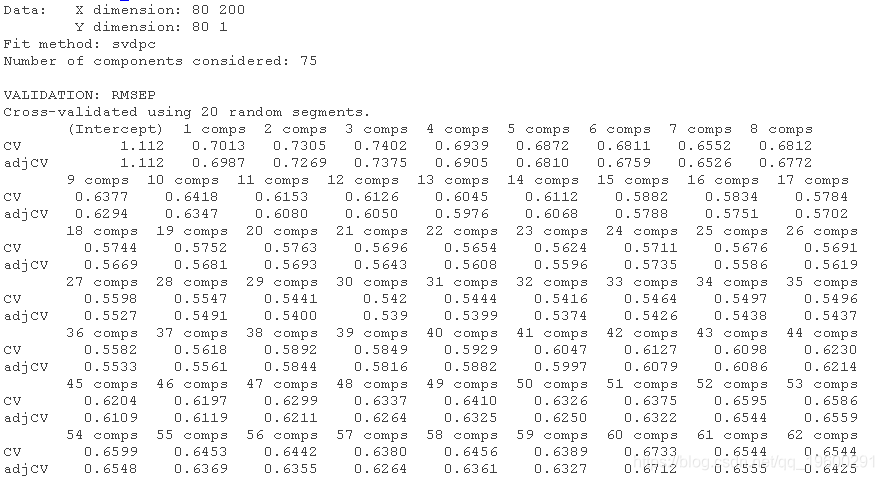
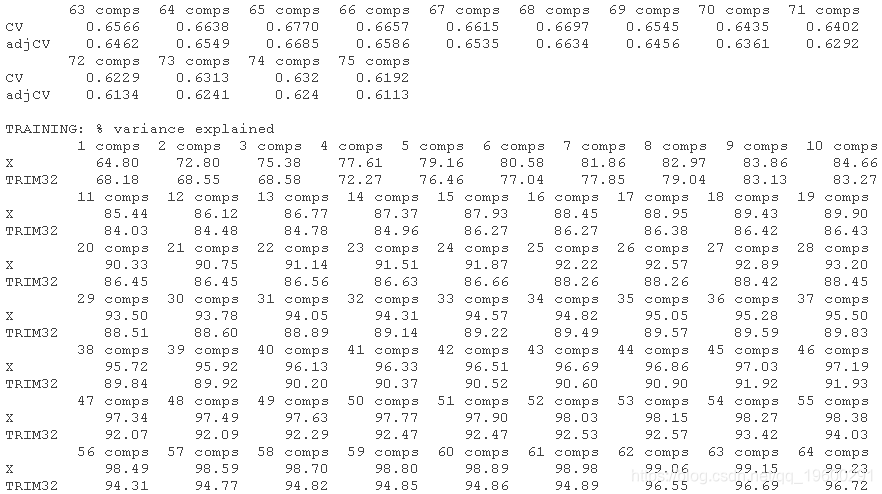

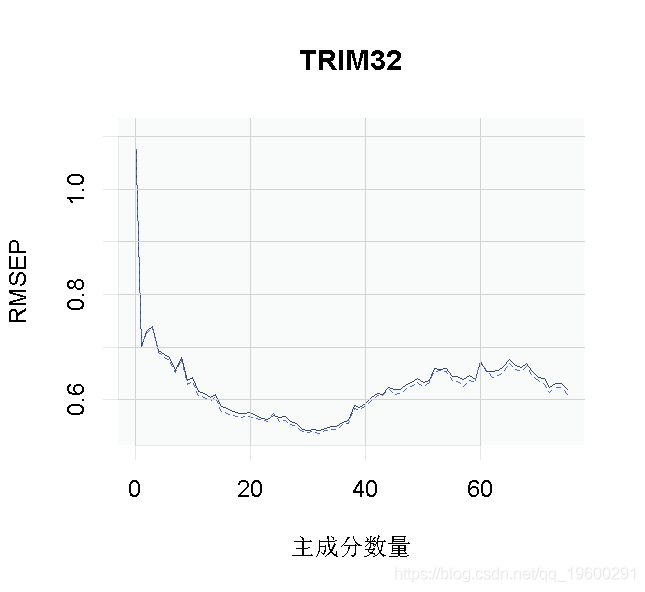
选择最佳的成分数。这里我们使用 "one-sigma "方法,它返回RMSE在绝对最小值的一个标准误差内的最低成分数。
plot(pcr, method = "onesigma")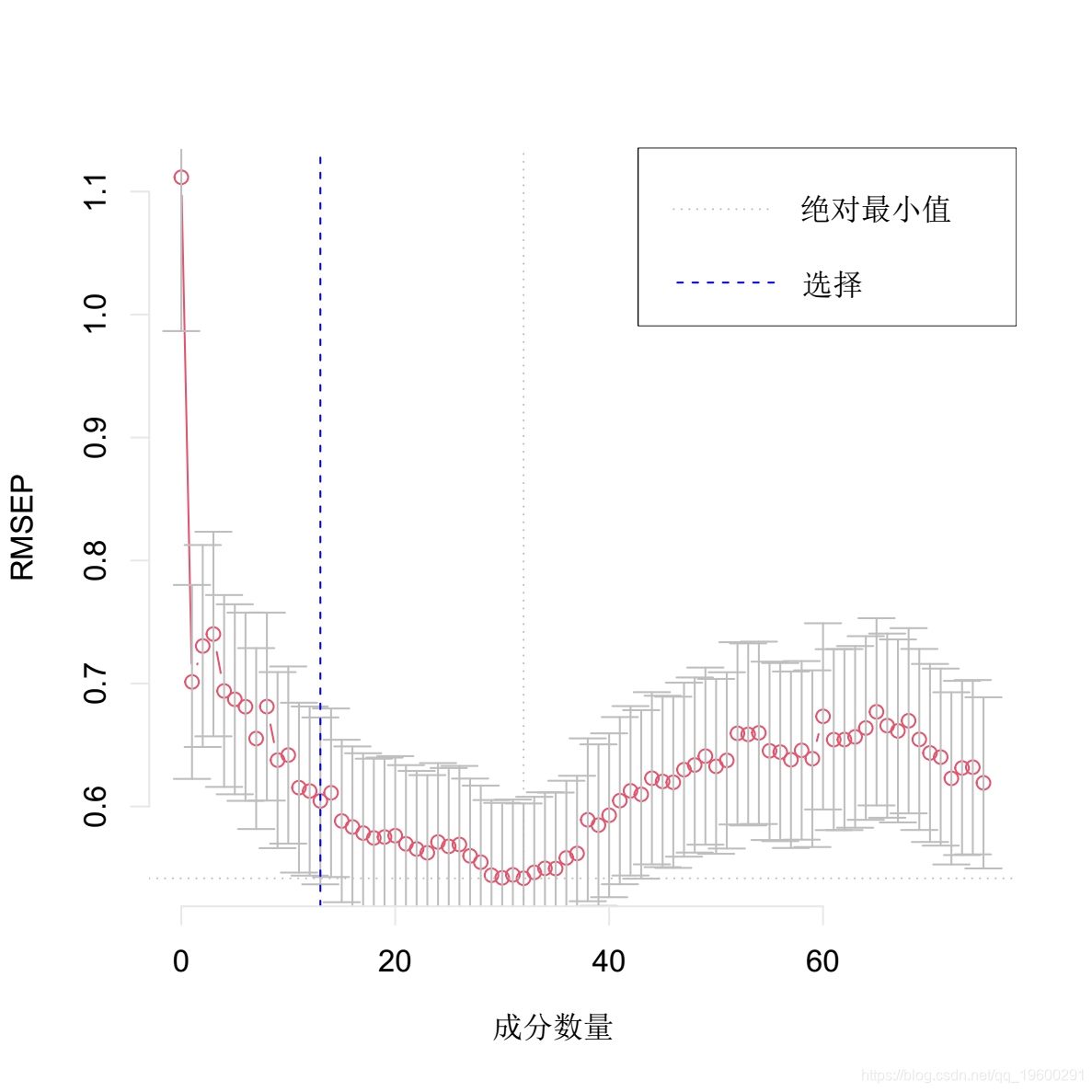
这个结果告诉我们,我们模型的最佳成分数是13。
8.2 对测试数据进行验证
我们现在使用最佳成分数来训练最终的PCR模型。然后通过对测试数据进行预测并计算MSE来验证这个模型。
我们定义了一个自定义函数来计算MSE。请注意,可以一次性完成预测和MSE计算。但是我们自己的函数在后面的lasso和ridge岭回归中会派上用场。
-
#平均平方误差
-
## obs: 观测值; pred: 预测值
-
MSE <- function(obs, pred)
-
pcr_preds <- predict(model, newdata = test_data, ncomp = optimal_ncomp)
-
![]()
这个值本身并不能告诉我们什么,但是我们可以用它来比较我们的PCR模型和其他类型的模型。
最后,我们将我们的因变量(TRIM32基因表达)的预测值与我们测试集的实际观察值进行对比。
plot(pcr_model, line = TRUE)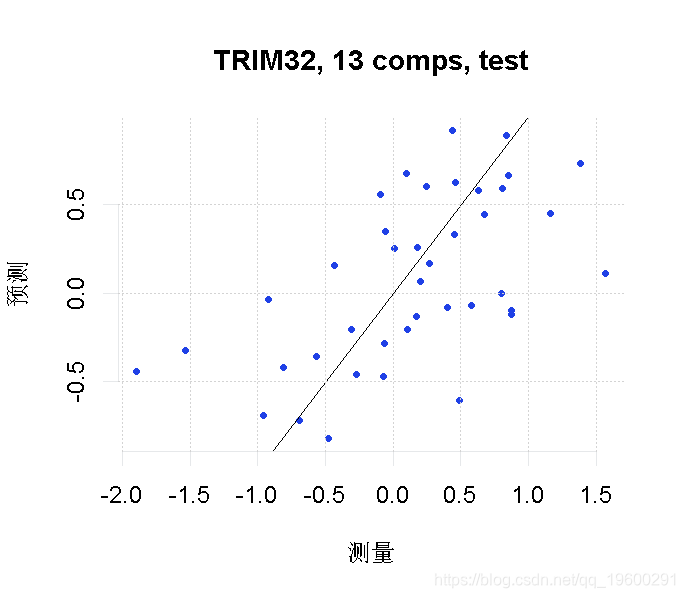
9 练习:评估和比较预测模型
1. 对训练数据(trainX, trainY)进行20次交叉验证的lasso回归。绘制结果并选择最佳的λ(γ)参数。用选定的lambda拟合一个最终模型,并在测试数据上验证它。
-
-
lasso_cv
-
#>
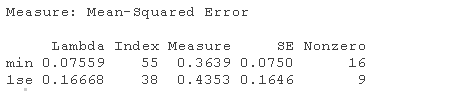

请注意,我们可以从CV结果中提取拟合的 lasso回归对象,并像以前一样制作系数曲线图。
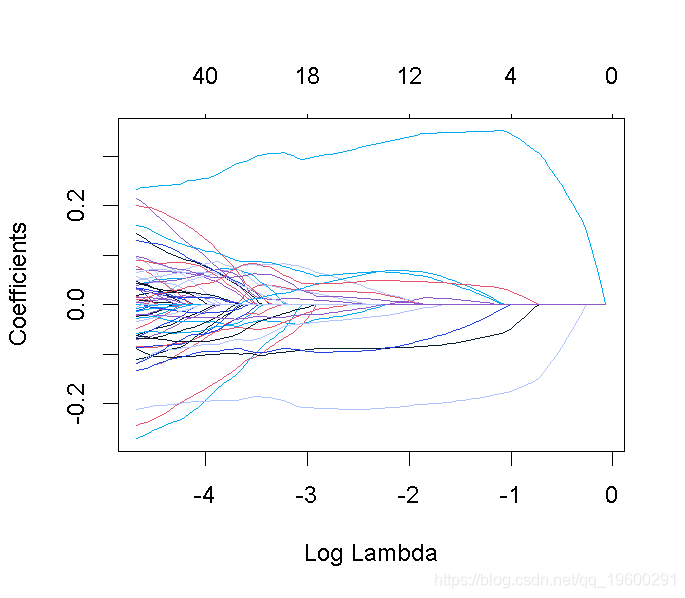
我们可以寻找能产生最佳效果的伽玛值。这里有两种可能性。
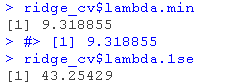
lambda.min: 给出交叉验证最佳结果的γ值。lambda.1se:γ的最大值,使MSE在交叉验证的最佳结果的1个标准误差之内。
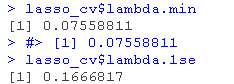
我们将在这里使用lambda.min来拟合最终模型,并在测试数据上生成预测。请注意,我们实际上不需要重新进行拟合,我们只需要使用我们现有的lasso_cv对象,它已经包含了lambda值范围的拟合模型。我们可以使用predict函数并指定s参数(在这种情况下设置lambda)来对测试数据进行预测。
![]()
2. 对岭回归做同样的处理。
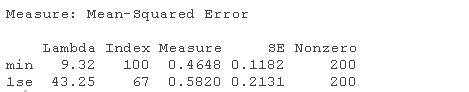
请注意,我们可以从CV结果中提取拟合的岭回归对象,并制作系数曲线图。
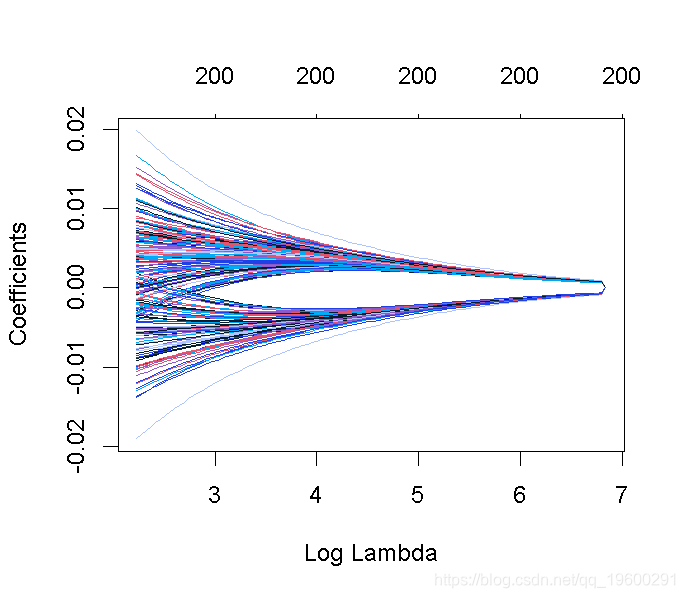
我们可以寻找能产生最佳效果的伽玛值。这里有两种可能性。
lambda.min: 给出交叉验证最佳结果的γ值。lambda.1se: γ的最大值,使MSE在交叉验证的最佳结果的1个标准误差之内。
我们在这里使用lambda.min来拟合最终的模型并在测试数据上生成预测。请注意,我们实际上不需要重新进行拟合,我们只需要使用我们现有的ridge_cv对象,它已经包含了lambda值范围的拟合模型。我们可以使用predict函数并指定s参数(在这种情况下混乱地设置lambda)来对测试数据进行预测。
-
ridge_preds <- predict
-
##计算MSE
![]()
3. 在所考虑的模型(PCR、lasso、岭回归)中,哪一个表现最好?
| 模型 | MSE |
|---|---|
| PCR | 0.3655052 |
| Lasso | 0.3754368 |
| Ridge | 0.3066121 |
-
注意:R中的log()默认是自然对数(以e为底),我们也会在文本中使用这个符号(比如上面图中的x轴标题)。这可能与你所习惯的符号(ln())不同。要在R中取不同基数的对数,你可以指定log的基数=参数,或者使用函数log10(x)和log2(x)分别代表基数10和2︎

最受欢迎的见解
3.matlab中的偏最小二乘回归(PLSR)和主成分回归(PCR)
5.R语言回归中的Hosmer-Lemeshow拟合优度检验