Single-molecule long-read sequencing facilitates shrimp transcriptome research 单分子长read测序促进了虾的转录组研究
Abstract
Although shrimp are of great economic importance, few full-length shrimp transcriptomes are available. Here, we used Pacific Biosciences single-molecule real-time (SMRT) long-read sequencing technology to generate transcripts from the Pacific white shrimp (Litopenaeus vannamei). We obtained 322,600 full-length non-chimeric reads, from which we generated 51,367 high-quality unique full-length transcripts. We corrected errors in the SMRT sequences by comparison with Illumina-produced short reads. We successfully annotated 81.72% of all unique SMRT transcripts against the NCBI non-redundant database, 58.63% against Swiss-Prot, 45.38% against Gene Ontology, 32.57% against Clusters of Orthologous Groups of proteins (COG), and 47.83% against Kyoto Encyclopedia of Genes and Genomes (KEGG) databases. Across all transcripts, we identified 3,958 long non-coding RNAs (lncRNAs) and 80,650 simple sequence repeats (SSRs). Our study provides a rich set of full-length cDNA sequences for L. vannamei, which will greatly facilitate shrimp transcriptome research.
Introduction
Whole-transcriptome analysis is of growing importance for animal biology research. However, whole-transcriptome analyses are ineffective without high quality transcript sequences1. Recently, second-generation sequencing (SGS) technologies, such as the Illumina Genome Analyzer, the Roche 454 pyrosequencing platform, and the ABI Solid platform, have facilitated the construction of transcriptome resources for many organisms2,3.
Shrimp are economically- and nutritionally-important crustaceans4. Several transcriptome studies in shrimp have been performed using SGS5, and many expressed sequence tags (ESTs) have been obtained6. However, the construction of transcriptomic sequences using SGS generally requires the assembly of short RNA-seq reads, and without a high-quality genome sequence available as a reference transcriptomic sequences may be misassembled due to reads transcribed from very similar members of multigene families or from highly repetitive regions7. In shrimp, the danger of misassembly may be even greater, as ~80% of the shrimp genome has been estimated to consist of repetitive elements8. Another limitation of SGS is that these technologies generally do not produce full-length transcripts, which are fundamental to studies of structural and functional genomics9,10,11. In addition, gene annotations and transcriptional characterizations of full-length transcripts are more accurate than those of transcript tags assembled from short RNA-sequencing reads7. Finally, alternative splicing, alternative polyadenylation, homologous genes, and superfamily genes are more easily identified based on full-length transcripts12,13,14,15.
Single-molecule real-time (SMRT) sequencing, a third-generation sequencing (TGS) technique recently developed by Pacific Biosciences (PacBio), allows direct sequencing of full-length, single-molecule cDNA sequences with a read length of up to 20 kb9,11,16. Using PacBio SMRT sequencing, intact RNA molecules can be sequenced without the need for fragmentation or post-sequencing assembly9. Thus, full-length transcripts can be constructed using SMRT sequencing.
The Pacific white shrimp (Litopenaeus vannamei) is the most extensively cultured crustacean species in the world, owing to its fast growth and strong disease resistance17,18. In this study, we used SMRT sequencing to construct the L. vannamei transcriptome. This is the first shrimp transcriptome constructed with SMRT.
摘要
虽然对虾具有很大的经济重要性,但很少有全长对虾转录组。
在这里,我们使用太平洋生物科学单分子实时(SMRT)长读测序技术从太平洋白虾(越南滨对虾)中生成转录本。
我们获得了322,600个全长非嵌合reads,从中我们获得了51,367个高质量的唯一全长转录本。
通过比较illumina产生的短片段,我们校正了SMRT序列中的错误。
在NCBI非冗余数据库中,我们成功注释了81.72%的所有独特的SMRT转录本,58.63%注释了Swiss-Prot, 45.38%注释了Gene Ontology, 32.57%注释了Orthologous Groups of proteins (COG), 47.83%注释了Kyoto Encyclopedia of Genes and Genomes (KEGG)数据库。
在所有的转录本中,我们鉴定出3958个长链非编码rna (lncRNAs)和80650个简单序列重复rna (SSRs)。
我们的研究为L. vannamei提供了一套丰富的全长cDNA序列,这将极大地促进对虾转录组的研究。
介绍
全转录组分析在动物生物学研究中越来越重要。
然而,如果没有高质量的转录序列,全转录组分析是无效的1。
最近,第二代测序(SGS)技术,如Illumina基因组分析仪、罗氏454焦磷酸测序平台和ABI Solid平台,促进了许多生物转录组资源的构建。
虾是一种经济上和营养上都很重要的甲壳动物。
利用SGS5对虾进行了多次转录组研究,并获得了许多表达序列标记(ESTs) 6。
然而,使用SGS构建转录组序列通常需要装配短RNA-seq reads,如果没有高质量的基因组序列作为参考,转录组序列可能会由于多基因家族中非常相似的成员或高度重复区域转录的reads而导致错误装配7。
在虾类中,错误组装的危险可能更大,因为据估计约80%的虾类基因组由重复元素组成8。
SGS的另一个局限性是,这些技术通常不能产生全长转录本,而全长转录本是结构和功能基因组研究的基础。
此外,基因注释和转录特征的全长转录比那些由短rna测序reads组装的转录子标签更准确7。
最后,选择性剪接、选择性聚腺苷酸化、同源基因和超家族基因更容易根据全长转录识别12,13,14,15。
单分子实时测序(SMRT)是太平洋生物科学(PacBio)最近开发的第三代测序(TGS)技术,可以直接测序全长、单分子cDNA序列,读取长度可达20 kb9、11、16。
使用PacBio SMRT测序,完整的RNA分子可以被测序,而不需要片段化或测序后组装。
因此,可以使用SMRT测序构建全长转录本。
太平洋白虾(凡纳滨对虾)生长迅速,抗病能力强,是世界上最广泛养殖的甲壳类动物。
在本研究中,我们使用SMRT测序构建了L.vannamei转录组。
这是第一个用SMRT构建的虾转录组。
Results
SMRT sequencing, quality filtering, and error correction
We used RNA extracted from six tissues (hepatopancreas, gills, heart, intestine, muscle, and stomach), collected and pooled from six L.vannamei, to constructed five cDNA libraries, each including cDNA inserts of approximately the same size: <1 kb, 1–2 kb, 2–3 kb, 3–6 kb, and >6 kb. We generated 1,307,853 polymerase reads (30.9 gigabases) across all five libraries. After removing adaptor sequences, low-quality sequences, and short sequences (<50 bp), 12,920,542 sub-reads remained. The mean sequence lengths for five cDNA libraries were 789 bp (<1 kb); 1,438 bp (1–2 kb); 2,304 bp (2–3 kb); 3,766 bp (3–6 kb); and 6,834 bp (>6 kb). We obtained 828,618 ROIs across all five cDNA libraries; the average lengths of the ROIs across the cDNA libraries were 2,018 bp, 2,968 bp, 3,340 bp, 4,235 bp, and 5,913 bp, respectively (Fig. 1). Of the 828,618 ROIs, 322,600 (38.93%) were identified as full-length non-chimeric (FLNC) reads.
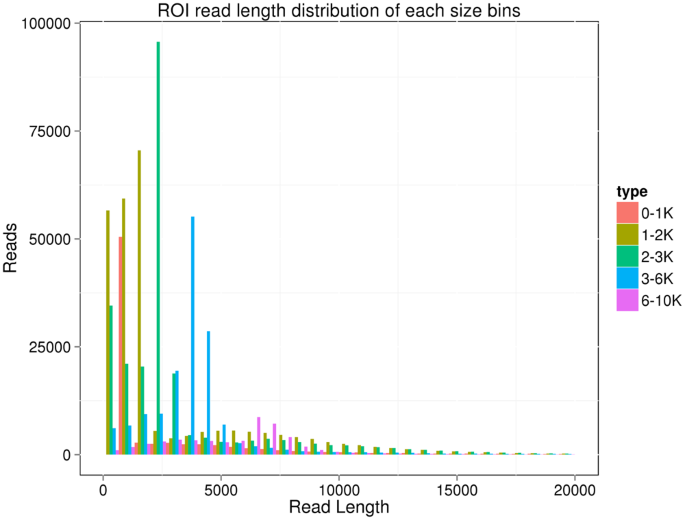
ROI read length distribution. Different colors represent different SMRT sequencing libraries with different cDNA insert size ranges.
We performed Illumina library construction and sequencing in parallel to correct the 322,600 FLNC reads. Using Illumina, ~148 million paired-end reads were sequenced, from which ~132 million clean reads were generated after adaptor sequence trimming and low-quality read filtering. We used Proovread19 to correct the FLNC reads based on the Illumina short reads. Proovread indicated that 124,201 FLNC reads (38.50%) contained at least one erroneous inner and/or terminal fragment; these fragments were corrected. We then used iterative clustering for error correction (IEC) to obtain 51,367 unique corrected SMRT transcripts.
To further test the completeness of our transcriptome, we used the Benchmarking Universal Single-Copy Orthologs (BUSCO) pipeline20 to compare our L. vannamei transcriptome to 1,066 conserved arthropod genes. This analysis indicated that 81.0% of the L. vannamei transcriptome (863 genes) encoded complete proteins. Of these genes, 34.3% (366 genes) were complete single-copy BUSCOs, 46.6% (497 genes) were complete duplicated BUSCOs, 3.1% (33 genes) were fragmented BUSCO archetypes, and 16.0% (170 genes) were missing BUSCOs entirely.
Functional annotation of transcripts
Of the 51,367 unique SMRT transcripts, we identified significant matches in the NCBI non-redundant (Nr) protein database for 41,975 (81.72%; E-value ≤ 10−5). Of the species with matches for >1.8% of all L. vannamei transcripts, 15.69% of the hits were from the termite Zootermopsis nevadensis, 9.81% were from L. vannamei, and 8.52% were from the crustacean Daphnia pulex (8.52%; Fig. 2).
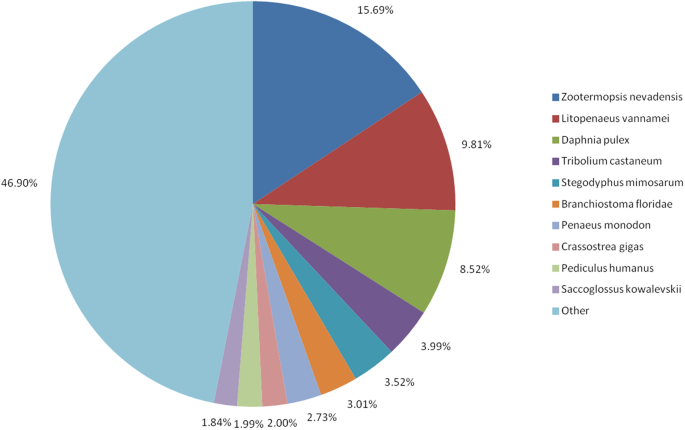
Percentage of L. vannamei transcripts with BlastX hits in various species. Transcripts were searched against the NCBI non-redundant protein database, using BlastX with the E-value cutoff set to <10−5. Only species with matches for >1.8% of the L. vannamei transcripts are shown; species matching fewer than 1.8% of all transcripts are classed as ‘Other’.
Our gene ontology (GO) analysis indicated that 9910 of the unique transcripts (42.51%) were enriched in biological processes, 8129 (34.87%) were enriched in molecular functions, and 5272 (22.62%) were enriched in cellular components (Fig. 3). We also identified matches to our unique transcripts in the Swiss-Prot21, Clusters of Orthologous Groups of proteins (COG)22, and Kyoto Encyclopedia of Genes and Genomes (KEGG)23 databases: 30,117 transcripts matched an entry in Swiss-Prot (58.63%), 16,732 transcripts matched an entry in COG (32.57%), and 24,569 transcripts matched an entry in KEGG (47.83%). The functional annotation of all unique transcripts are listed in Supplementary Table 1.
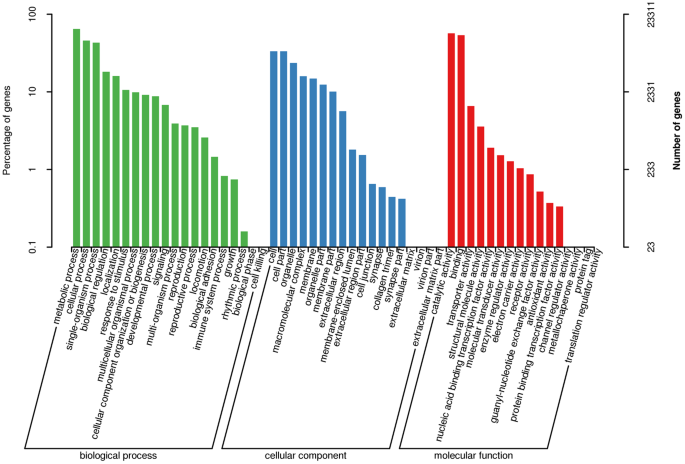
GO classification of the putative functions of the unique transcripts of L. vannamei.
To further identify the protein coding potential of unique transcripts, we predicted ORFs within all unique transcripts. In total, 47,260 unique transcripts were found having the protein coding potential, with an average length of 3,493 bp. The length distribution indicated that most protein-coding unique transcripts were distributed in length from 300 bp to 1,0000 bp, and there were more than 600 transcripts with a length >10,000 bp. (Fig. 4).
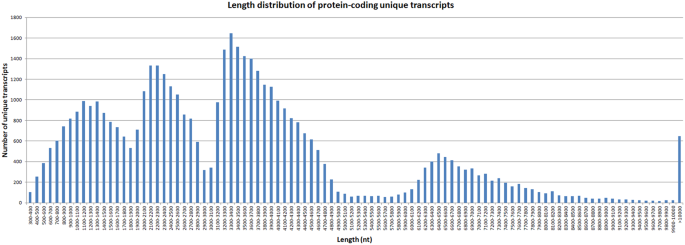
Lengths of candidate protein-coding RNAs.
Identification of long non-coding RNAs (lncRNAs)
We used four tools to identity unique transcripts without protein coding potential (i.e., lncRNAs): the Coding Potential Calculator (CPC)24 identified 375 lncRNAs, the Coding-Non-Coding Index (CNCI)25 identified 2,178 lncRNAs, the Coding Potential Assessment Tool (CPAT)26 identified 751 lncRNAs, and Pfam27 identified 4,342 lncRNAs. In total, 5893 unique transcripts were identified as lncRNAs by at least one tool (Fig. 5). After candidate lncRNAs with EMBOSS-predicted ORFs > 100 bp were removed, 3,958 lncRNAs remained. The average length of these lncRNAs was 2,111 bp, with most lncRNAs ranging in length from 300 bp to 4,800 bp (Fig. 6).
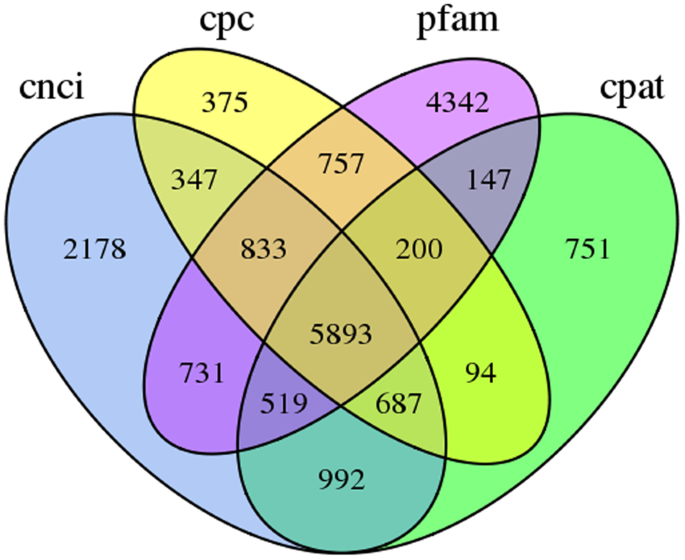
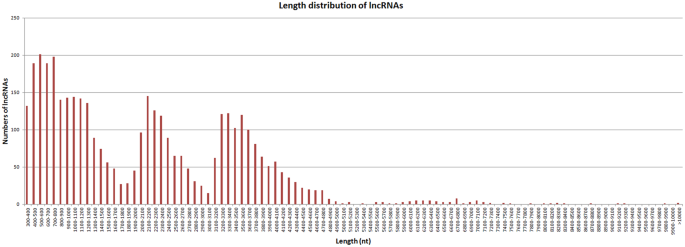
Lengths of candidate lncRNAs.
Identification of simple sequence repeats (SSRs)
SSRs are repetitive sequence motifs approximately 1–6 bp long28. We searched for SSRs in the 50,688 unique L. vannamei transcripts longer than 500 bp. We identified 80,650 SSRs across all tested transcripts, with 17,222 (33.98%) unique transcripts containing more than one SSR. Most of the SSRs identified were mono-nucleotide repeats (50.81%), followed by the di-nucleotide repeats (27.55%), tri-nucleotide repeats (18.33%), tetra-nucleotide repeats (2.41%), hexa-nucleotide repeats (0.55%), and penta-nucleotide repeats (0.35%). All SSRs and their primers are listed in Supplementary Table 2.
Comparison with previous L.vannamei transcriptomes
Strikingly, most of the assembled unique transcripts generated by Illumina and 454 sequencing were <1000 bp in length, while the lengths of the SMRT assembled unique transcripts were much more evenly distributed, with a considerable proportion of assembled transcripts ~6000–8000 bp long (Fig. 7). With respect to transcript functional annotations, proportionally more SMRT-sequenced transcripts were annotated than either 454-pyrosequenced transcripts or Illumina-sequenced transcripts (Fig. 8).
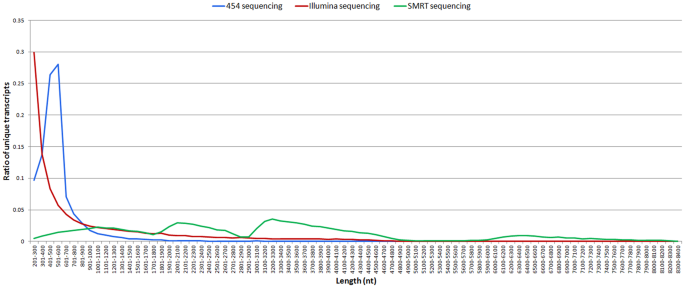
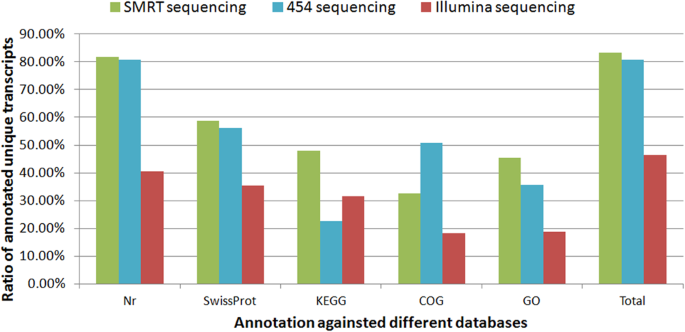
Discussion
Full-length cDNA sequences are useful for functional studies of important genes. However, full-length cDNA sequences can often only be generated by rapid amplification of cDNA ends (RACE), which is time consuming, labor intensive, expensive, and inefficient29. To date, very few full-length cDNA sequences have been reported for shrimp. Here, we used PacBio SMRT sequencing to obtain 51,367 high-quality unique full-length transcripts for L. vannamei. This large number of full-length cDNA sequences will greatly facilitate research projects using the shrimp transcriptome.
We compared several previously reported full-length cDNAs from L. vannamei with the corresponding full-length transcripts obtained in this study, including C-type lectin30, prophenoloxidase31, and ferritin32. We found the SMRT transcripts were essentially identical to the RACE cDNAs, with only minor differences at the 5′ and 3′ ends. These differences might have been due to differences in the primer sequences used by SMRT and RACE. Thus, our results suggested that SMRT sequencing is an effective method by which to obtain full-length cDNA sequences from the shrimp transcriptome.
Short-read sequencing (Illumina or 454) has been used to produce transcriptomes of some shrimp species, including L. vannamei17,18,33,34,35,36,37,38,39,40, Fenneropenaeus merguiensis41,42, Macrobrachium rosenbergii43, Triops newberryi44, T. longicaudatus45, Pandalus latirostris46, Fenneropenaeus chinensis47, Palaemon serratus48, and Penaeus monodon49. The average lengths of transcripts obtained in these studies were ~306–1,027 bp. Here, the average length of SMRT-sequenced transcripts was nearly 3 kb, far exceeding those of the previous studies. Our findings thus indicated that long transcripts in shrimp, from both coding and non-coding genes, might be more prevalent than previously estimated33.
Although SMRT sequencing produces longer reads than SGS methods, the SMRT raw data error rate is relatively high50. To correct these errors, it is possible to use the short reads generated by SGS as references51,52. Here, we used Illumina sequences to correct the SMRT reads. As 38.50% of the SMRT FLNC reads contained erroneous fragments (or single-nucleotide bases), our results indicated that error correction processing should be performed before further analysis of SMRT sequences.
LncRNAs are non-coding RNAs that are longer than 200 nucleotides long53,54. LncRNAs evolve rapidly, and are often species-specific in plants or animals55. An accumulating body of evidence has suggested that lncRNAs play essential roles in many important biological processes, such as translation, transcription, differentiation, splicing, immune responses, epigenetic regulation, and cell cycle control54,56,57,58,59. However, no lncRNAs in crustaceans have previously been reported. Here, we identified 3,958 novel lncRNAs in the L. vannamei shrimp transcriptome. These newly identified lncRNAs will be useful for several aspects of shrimp research, including epigenetics, immunology, and phylogenomics.
The SMRT transcriptome obtained here had a longer average transcript length than the transcripts obtained with SGS. Our results suggested that full-length transcripts were more easily annotated than shorter transcripts. Here, 81.72% of unique transcripts were annotated in the Nr database, as compared to 37.80%–73.08% in previously published L. vannamei transcriptomes produced with short-read sequencing17,18,33,34. This suggested that full-length transcripts were annotated more efficiently than the ESTs obtained by assembling short RNA-sequence reads.
全长cDNA序列对于重要基因的功能研究是有用的。
然而,cDNA全长序列往往只能通过cDNA末端(RACE)的快速扩增而产生,这是费时、劳动密集型、昂贵且低效的。
到目前为止,很少有虾的全长cDNA序列被报道。
在这里,我们使用PacBio SMRT测序获得了51367个高质量的vannamei唯一全长转录本。
大量的全长cDNA序列将极大地促进利用虾类转录组的研究项目。
我们比较了先前报道的几种来自L. vannamei的全长cDNAs与本研究中获得的相应全长转录本,包括c型凝集素30、prophenoloxidase31和ferritin32。
我们发现,SMRT转录本与RACE cDNAs基本相同,只有在第5和第3端有微小差异。
这些差异可能是由于SMRT和RACE使用的引物序列不同造成的。
因此,我们的结果表明,SMRT测序是一种有效的方法,可以从虾的转录组中获得全长cDNA序列。
短内容排序(Illumina公司或454)已经被用来生产一些虾物种的转录组,包括l . vannamei17 18岁,33、34、35、36、37、38、39、40岁Fenneropenaeus merguiensis41, 42岁,Macrobrachium rosenbergii43,蟹newberryi44, t . longicaudatus45 Pandalus latirostris46, Fenneropenaeus chinensis47, Palaemon serratus48,和中国对虾monodon49。
在这些研究中获得的转录子的平均长度约为306 1027 bp。
这里,smrt测序的平均长度接近3 kb,远远超过了之前的研究。
因此,我们的发现表明,在虾中,编码和非编码基因的长转录本可能比之前估计的更普遍。
虽然SMRT测序比SGS方法产生更长的读取,但SMRT原始数据错误率相对较高。
为了纠正这些错误,可以使用SGS产生的短读取作为参考51、52。
在这里,我们使用Illumina序列来校正SMRT的读取。
由于38.50%的SMRT FLNC reads含有错误片段(或单核苷酸碱基),我们的结果表明,在进一步分析SMRT序列之前,需要进行错误校正处理。
lncrna是非编码rna,长度超过200个核苷酸53,54。
lncrna进化迅速,在植物或动物中通常具有物种特异性55。
越来越多的证据表明,lncrna在许多重要的生物学过程中发挥着重要的作用,如翻译、转录、分化、剪接、免疫反应、表观遗传调控和细胞周期控制54,56,57,585,59。
然而,在甲壳类动物中尚未见lncrna的报道。
在这里,我们在L. vannamei虾转录组中鉴定了3958个新的lncrna。
这些新发现的lncrna将在对虾研究的几个方面有用,包括表观遗传学、免疫学和系统基因组学。
这里获得的SMRT转录组的平均转录本长度比SGS获得的转录本长。
我们的结果表明,全长的转录比短的转录更容易被注释。
在本研究中,81.72%的独特转录本被注释在Nr数据库中,而在先前发表的序列为17、18、33、34的L. vannamei转录组中,这一比例为37.80% 73.08%。
这表明,与通过组装短rna序列片段获得的est相比,对全长转录本进行注释的效率更高。
Materials and Methods
Animal materials
Specific pathogen-free (SPF) white shrimp (L. vannamei) were obtained from the National and Guangxi Shrimp Genetic Breeding Center (Guangxi Province, China). We removed and pooled the hepatopancreases, gills, hearts, intestines, muscles, and stomachs of six shrimp. Pooled tissues were immediately stored in liquid nitrogen until RNA extraction.
RNA extraction
Total RNA was extracted from the pooled tissues using TRIzol LS Reagent (Invitrogen, USA) following the manufacturer’s instructions, and genomic DNA was removed using DNase I (Invitrogen, USA). RNA purity (OD260/280), concentration, and absorption peak were measured using a NanoDrop 2000 (Thermo Scientific, USA). RNA quality was determined with a Bioanalyser 2100 (Agilent, USA). Only total RNAs with a RIN score >7 were used to construct cDNA libraries for SMRT sequencing.
SMRT library construction, sequencing, and quality control
To construct full-length cDNAs, 10 μg of total RNA was reverse transcribed into cDNA using a SMARTer PCR cDNA Synthesis Kit (Takara, Japan), following the manufacturer’s protocols. Size fractionation and selection were performed using the BluePippin Size Selection System (Sage Science, USA). We prepared five SMRT libraries, each including fragments in one of five size groups: <1 kb, 1–2 kb, 2–3 kb, 3–6 kb, and >6 kb, following the PacBio protocol. Each library was sequenced in three SMRT cells on a PacBio RSII platform using C4 reagents and 3–4 h sequencing movies.
We used PacBio SMRT analysis software v2.3.0 (http://www.pacb.com/products-andservices/analytical-software/smrt-analysis/) to filter out low-quality polymerase reads (read-length <50 bp and read-score <0.75). ROIs were filtered from the sub-reads with the full pass threshold set to ≥0 and the predicted unique accuracy set to ≥0.75. We considered ROIs FLNC reads only if they possessed a 5′-cDNA primer, a 3′-cDNA primer, and a polyA tail preceding the 3′ primer. Then 5′- and 3′-cDNA primers and polyA tail were removed from FLNC according to the Pac-bio recommended procedure (https://github.com/PacificBiosciences/IsoSeq.3).
Illumina library construction and sequencing
The Illumina libraries used to correct the FLNC reads were constructed with the Tru-Seq RNA sample Prep kit (Illumina, USA). Briefly, poly-(A) mRNA was isolated from total RNA using oligo (dT) magnetic beads and then fragmented into 200–700 bp pieces with fragmentation buffer. Double-stranded cDNAs were synthesized using a SuperScript double-stranded cDNA synthesis kit (Invitrogen, USA) with random hexamer primers (Illumina, USA), following the manufacturer’s instructions. Synthesized cDNAs were gen-purified and amplified with PCR. PCR products were sequenced on a single lane of an Illumina HiSeq. 2500 high-throughput sequencer. Raw sequencing reads were quality controlled to remove adaptor sequences, low-quality reads (reads where quality was ≤10% for >50% of all nucleotides), and read with many unknown nucleotides (>10%). Cleaned sequences were used for SMRT error correction.
Quality filtering and error correction of PacBio reads
Nucleotide errors in the FLNC reads were corrected by comparison with the Illumina RNA sequences using Proovread v2.13.13 (https://github.com/BioInf-Wuerzburg/proovread) with parameter coverage set to 507,19. Corrected FLNC reads were clustered into unique (non-redundant) transcripts using the ICE algorithm in the PacBio SMRT analysis software v2.3.0, with quiver polishing set to ≥0.9955,60. We used BUSCO v3.0 (http://busco.ezlab.org/)20 with the BUSCO arthropod dataset (http://busco.ezlab.org/v2/datasets/arthropoda_odb9.tar.gz) to evaluate the completeness of the L. vannamei transcriptome.
Functional annotation of transcripts
We identified functional annotations matching each unique transcript by searching Nr, Swiss-Prot, COG, and KEGG using BlastX with an E-value cut-off of 10−5. Protein function was predicted based on the annotation of the most similar hit across all databases. The unique transcripts identified by BlastX were submitted to blast2GO v4.1 (http://www.blast2go.com)61 to assign GO categories. To identify the protein coding potential of each unique transcript, the ORFs within unique transcripts were predicted using TransDecoder v2.0.1 (https://transdecoder.github.io)62, with default parameters.
Identification of lncRNAs
We identified unique transcripts without protein coding potential as candidate lncRNAs using four tools: CPC v1.0 (http://cpc.cbi.pku.edu.cn/)24, CNCI v2.0 (https://github.com/www-bioinfo-org/CNCI)25, CPAT v1.2 (http://lilab.research.bcm.edu/cpat/index.php)26, and Pfam (http://pfam.xfam.org/)27 with default parameters. We then predicted the ORFs of all candidate lncRNAs selected by at least one tool with EMBOSS getorf v6.1.063; sequences containing ORFs > 100 bp long were discarded.
Identification of SSRs
We used MISA v1.0 (http://pgrc.ipk-gatersleben.de/misa/)64 with default parameters to identify SSRs (mono- to penta-nucleotide repeats) in all corrected unique transcripts longer than 500 bp. SSR primers were designed using primer365 with default parameters.
Comparison with previously published L. vannamei transcriptomes
To evaluate SMRT sequencing performance, we compared the SMRT transcriptome constructed here to two previously published L. vannamei transcriptomes, one obtained using 454 sequencing17 and one obtained using Illumina sequencing18. First, we compared the distributions of transcript lengths among the three transcriptomes. Next, we compared the number of Nr, Swiss-Prot, KEGG, COG and GO hits among the transcriptomes (all functional annotations for each of the three transcriptomes were performed with an E-value cutoff of 10−5).
动物材料
SPF白虾(L. vannamei)从国家和广西对虾遗传育种中心(中国广西)获得。
我们切除并合并了六只虾的肝皱褶、鳃、心脏、肠子、肌肉和胃。
汇集的组织立即保存在液氮中直到RNA提取。
RNA提取
按照制造商的说明,使用TRIzol LS试剂(Invitrogen公司,美国)从合并组织中提取总RNA,使用DNase I去除基因组DNA (Invitrogen公司,美国)。
使用NanoDrop 2000 (Thermo Scientific, USA)测定RNA纯度(OD260/280)、浓度和吸收峰。
使用生物分析仪2100(安捷伦,美国)测定RNA质量。
只有RIN评分为7的总rna被用于构建用于SMRT测序的cDNA文库。
SMRT库的构建、测序和质量控制
为了构建全长cDNAs,使用更智能的PCR cDNA合成试剂盒(Takara,日本),按照制造商的协议,将总RNA的10立方克逆转录为cDNA。
采用蓝epippin粒度选择系统(Sage Science, USA)进行粒度分选。
按照PacBio协议,我们准备了5个SMRT库,每个库包括5个大小组中的片段:1 kb、1 - 2 kb、2-3 kb、3-6 kb和6 kb。
每个文库在PacBio RSII平台上的三个SMRT细胞中使用C4试剂和3-4 h测序膜进行测序。
我们使用PacBio SMRT分析软件v2.3.0 (http://www.pacb.com/products-andservices/analytical-software/smrt-analysis/)过滤掉低质量的聚合酶reads(读取长度<50 bp,读取分数<0.75)。
从子reads中过滤roi,并将全通阈值设置为≥0,预测唯一精度设置为≥0.75。
我们认为ROIs FLNC只有具备5 ' -cDNA引物、3 ' -cDNA引物和3 '引物前的polyA尾巴时才会读取。
然后按照Pac-bio推荐程序(https://github.com/pacificbiosciences/isoseq3)从FLNC中取出5 ' -和3 ' - cdna引物和polyA尾巴。
Illumina图书馆的构建和测序
用于修正FLNC reads的Illumina文库由truo - seq RNA样品准备试剂盒(Illumina,美国)构建。
简单地说,使用oligo (dT)磁珠从总RNA中分离出poly-(A) mRNA,然后使用fragmentation buffer将其裂解成200-700 bp的片段。
使用上标双链cDNA合成试剂盒(Invitrogen公司,美国),使用随机六聚体引物(Illumina公司,美国),按照制造商的说明合成双链cDNAs。
对合成的cDNAs进行基因纯化,PCR扩增。
PCR产物在Illumina HiSeq单车道上进行测序。
2500高通量测序仪。
对原始测序reads进行质量控制,以去除接头序列、低质量的reads(50%核苷酸质量≤10%的reads)和含有许多未知核苷酸的reads (>10%)。
清洗后的序列用于SMRT纠错。
PacBio读取的质量过滤和纠错
使用Proovread v2.13.13 (https://github.com/BioInf-Wuerzburg/proovread)对FLNC reads中的核苷酸错误与Illumina RNA序列进行比较,参数覆盖范围设置为507,19。
使用PacBio SMRT分析软件v2.3.0中的ICE算法将校正后的FLNC读取聚类为唯一(非冗余)转录本,quiver抛光设置为≥0.9955,60。
我们使用BUSCO v3.0 (http://busco ezlab.org/)20和BUSCO arthropod数据集(http://busco ezlab.org/v2/datasets/arthropoda_odb9.tar.gz)来评估L. vannamei转录组的完整性。
转录本的功能注释
通过使用E-value cut- 10 - 5的BlastX搜索Nr、Swiss-Prot、COG和KEGG,我们确定了匹配每个独特转录本的功能注释。
蛋白质功能的预测是基于所有数据库中最相似的击中的注释。
BlastX鉴定出的唯一转录本被提交到blast2GO v4.1 (http://www.blast2go.com)61来划分GO类别。
为了识别每个独特转录本的蛋白编码潜力,使用TransDecoder v2.0.1 (https://transdecoder.github.io)62,使用默认参数预测独特转录本中的ORFs。
lncRNAs的识别
利用四种潜在的crv1.0编码工具确定lncpc为唯一的候选蛋白(http://cpc.cbi.pku.edu.cn/)24,中色国际v2.0(https://github.com/www-bioinfo-org/CNCI)25,CPAT 1.2版(http://lilab.research.bcm.edu/cpat/index.php)26和Pfam(http://pfam.xfam.org/)27,默认参数。然后,我们用浮雕getorf v6.1.063预测了至少一个工具选择的所有候选lncrna的orf;包含长度为 > 100 bp的序列被丢弃。
ssr的鉴定
我们使用MISA v1.0(http://pgrc.ipk-gatersleben.de/misa/)64使用默认参数来识别所有校正过的长度超过500 bp的独特转录物中的SSR(单核苷酸到五核苷酸重复序列)。SSR引物是用primer365设计的,带有默认参数。
与先前发表的凡纳霉转录本比较
为了评估SMRT测序性能,我们将这里构建的SMRT转录组与两个先前发表的凡纳麦L.vannamei转录组进行了比较,一个使用454序列17获得,另一个使用Illumina序列18获得。首先,我们比较了三个转录体之间转录长度的分布。接下来,我们比较了转录组中Nr、Swiss Prot、KEGG、COG和GO的点击数(三个转录体中的每一个都执行了功能注释,E值截止值为10-5)。
Data Availability
Raw PacBio sequencing reads are available at NCBI GenBank under the accession SRX3267788, SRX3267789, SRX3267790, SRX3267791, SRX3267792, SRX3267793, SRX3267794, SRX3267795, SRX3267796, SRX3267797, SRX3267798, SRX3267799, SRX3267800, and SRX3267801). Raw Illumina sequencing reads are available at NCBI GenBank under the accession SRX3527198 and SRX3527197. Candidate protein-coding transcripts are available at NCBI GenBank under the accession GGUK00000000. Candidate lncRNA sequences are available at NCBI GenBank under the accession GGUT00000000.