处理基因组数据,很多时候我们会觉得直接看序列文件不够直观,如果绘图的话,把n多G把数据用画图出来不仅费劲,就算操作也不方便。因此我们可以用UCSC开发出的genome browser,可以直接把数据信息写成track,连上genome browser 上查看,它还支持安装到本地服务器上(genome browser in box ,简称GBIB),genome browser 支持的格式有bedGraph, GTF, PSL, BED, bigBed, WIG, bigGenePred, bigMaf, bigChain, bigPsl, bigWig, BAM, CRAM, VCF, MAF, BED detail, Personal Genome SNP, broadPeak, narrowPeak, and microarray (BED15),GFF和GTF文件必须tab分隔。 废话少说,直接入门。本文主要讲SAM,BAM,WIG,bigWig,VCF,BED文件上传及使用。
一、格式的前期处理
1.1 WIG 和 bigWig
WIG 文件格式,有两种可选的格式,variableStep和fixedStep。variableStep用于区间变化的,fixedStep用于区间固定的。
variableStep WIG文件以variableStep 开头,chrom染色体,可选参数span(默认span=1),指定每一行的位置区间,比如2,区间就是chromStart~chromStart+2。chromStart染色体位置,dataValue染色体位置上的值。
1 variableStep chrom=chrN 2 [span=windowSize] 3 chromStartA dataValueA 4 chromStartB dataValueB 5 ... etc ... ... etc ...
fixedStep文件以fixedStep开头,chrom染色体,start是起始固定的位置,step是每两个起始position之间的间隔,span和variableStep中的step一样,指定每一行的位置区间。
这样dataValue1对应的position是start~start+span,dataValue2对应的position是start+step~start+step+span.
1 fixedStep chrom=chrN 2 start=position step=stepInterval 3 [span=windowSize] 4 dataValue1 5 dataValue2 6 ... etc ...
WIG格式要在genome browser 上查看最好转换为bigWig文件,bigWig文件是index后的二进制WIG文件,在genome browser上查看更加快速,用wigToBigWig命令
1 wigToBigWig sample.wig chrom.sizes output.bw
chromsizes 文件可以从UCSC上下载,就是各个染色体的长度大小hg19.chrom.sizes可以从这里直接复制。


1 chr1 249250621 2 chr2 243199373 3 chr3 198022430 4 chr4 191154276 5 chr5 180915260 6 chr6 171115067 7 chr7 159138663 8 chrX 155270560 9 chr8 146364022 10 chr9 141213431 11 chr10 135534747 12 chr11 135006516 13 chr12 133851895 14 chr13 115169878 15 chr14 107349540 16 chr15 102531392 17 chr16 90354753 18 chr17 81195210 19 chr18 78077248 20 chr20 63025520 21 chrY 59373566 22 chr19 59128983 23 chr22 51304566 24 chr21 48129895 25 chr6_ssto_hap7 4928567 26 chr6_mcf_hap5 4833398 27 chr6_cox_hap2 4795371 28 chr6_mann_hap4 4683263 29 chr6_apd_hap1 4622290 30 chr6_qbl_hap6 4611984 31 chr6_dbb_hap3 4610396 32 chr17_ctg5_hap1 1680828 33 chr4_ctg9_hap1 590426 34 chr1_gl000192_random 547496 35 chrUn_gl000225 211173 36 chr4_gl000194_random 191469 37 chr4_gl000193_random 189789 38 chr9_gl000200_random 187035 39 chrUn_gl000222 186861 40 chrUn_gl000212 186858 41 chr7_gl000195_random 182896 42 chrUn_gl000223 180455 43 chrUn_gl000224 179693 44 chrUn_gl000219 179198 45 chr17_gl000205_random 174588 46 chrUn_gl000215 172545 47 chrUn_gl000216 172294 48 chrUn_gl000217 172149 49 chr9_gl000199_random 169874 50 chrUn_gl000211 166566 51 chrUn_gl000213 164239 52 chrUn_gl000220 161802 53 chrUn_gl000218 161147 54 chr19_gl000209_random 159169 55 chrUn_gl000221 155397 56 chrUn_gl000214 137718 57 chrUn_gl000228 129120 58 chrUn_gl000227 128374 59 chr1_gl000191_random 106433 60 chr19_gl000208_random 92689 61 chr9_gl000198_random 90085 62 chr17_gl000204_random 81310 63 chrUn_gl000233 45941 64 chrUn_gl000237 45867 65 chrUn_gl000230 43691 66 chrUn_gl000242 43523 67 chrUn_gl000243 43341 68 chrUn_gl000241 42152 69 chrUn_gl000236 41934 70 chrUn_gl000240 41933 71 chr17_gl000206_random 41001 72 chrUn_gl000232 40652 73 chrUn_gl000234 40531 74 chr11_gl000202_random 40103 75 chrUn_gl000238 39939 76 chrUn_gl000244 39929 77 chrUn_gl000248 39786 78 chr8_gl000196_random 38914 79 chrUn_gl000249 38502 80 chrUn_gl000246 38154 81 chr17_gl000203_random 37498 82 chr8_gl000197_random 37175 83 chrUn_gl000245 36651 84 chrUn_gl000247 36422 85 chr9_gl000201_random 36148 86 chrUn_gl000235 34474 87 chrUn_gl000239 33824 88 chr21_gl000210_random 27682 89 chrUn_gl000231 27386 90 chrUn_gl000229 19913 91 chrM 16571 92 chrUn_gl000226 15008 93 chr18_gl000207_random 4262
1.2 sam 和 bam文件
sam/bam 格式是mapping后的序列比对文件,sam文件需要先转成bam,sam/bam文件传到genome browser上可以看到reads在chrom上的分布。bam文件需要sort后建立index,并且要将index 文件*.bai放到bam文件所在目录下。
如果是sam 文件,先转变为bam文件
1 samtools view -S -b -o sample.bam sample
进行sort,并且建立index
1 samtools sort sample.bam sample.sorted 2 samtools index sample.sorted.bam
1.3 VCF文件
vcf 文件是千人基因组计划发展出的存储基因组变异信息的文件,包括SNP和结构变异信息。传到genome browser上可以看到不同位点的变异信息。
先要对vcf 格式就行sort,用vcftools 中的vcf-sort,没有的话需要去下载,https://sourceforge.net/projects/vcftools/
1 vcf-sort sample.vcf > sample.sorted.vcf
要下载bgzip 和 tabix 程序,https://sourceforge.net/projects/samtools/files/tabix/.
对sort后的vcf 进行压缩
1 bgzip sample.sorted.vcf sample.sorted.vcf.gz
对vcf.gz文件建立index
1 tabix -p vcf sample.sorted.vcf.gz
建立track的时候,要把tbi格式的index放在vcf.gz所在的文件夹下。
1.4 bed和bigBed文件
1.4.1 bed文件格式
1.4.1.1 必须的三个区域:
1.chrom 染色体
2.chromStart 在染色体上的起始位置
3.chromEnd 在染色体上的结束位置
1.4.1.2有九个额外的可选的区域
4.name 行名
5.score 分值 0-1000,影响显示的灰色深度
6.strand 正负链,"."无方向,或者“+”或者"-"
7.thickStart 开始浓密绘制的位置
8.thickEnd 结束浓密绘制的位置
9.itemRgb RGB值,R、G、B值(比如255,0,0),如果itemRgb属性设置为开的话,RGB将设置这一行的颜色
10.blockCount 该行的区块(外显子)数目
11.blockSizes 逗号分隔的区块大小的列表,
12.blockStarts 逗号分隔的区块开始位置,所以的区块开始位置都应该能由chromStart计算出来,位置数目应该与blockSizes数目相裂隙。
bed文件可以在前面添加track和browser行,作为一个track传上genome browser。后面会详细说明。
1.4.2 bigBed文件
如果bed文件有点大(大于50Mb),你应该将它转换成bigBed文件,放到服务器上,再链接到genome browser上查看。
先sort bed文件
1 bedSort unsorted.bed > input.bed
将sort后的bed文件进行转换,必须去除track和browser行
1 bedToBigBed input.bed chrom.sizes myBigBed.bb
二、在UCSC上查看数据
2.1 UCSC 上My Data 下的Custom Track
所有文件都可以直接添加自己定制的Custom Track,分为两步,1.定义browser行 ,2.定义track行
1.browser行
1 browser attribute_name attribute_value(s)
postion 定义genome browser起始查看的位置
hide all 隐藏全部track
hide < track_primary_talbe_name(s)> 需要隐藏的tracks列表,空格分隔,下面一样
dense all 密度显示全部track
dense <track_primary_talbe_name(s)> 需要密度显示的tracks列表
pack all 压紧模式显示全部track
pack <track_primary_talbe_name(s)> 需要压紧模式显示的tracks列表
squish all 压扁模式显示
full all 全部显示track
full <track_primary_talbe_name(s)> 全部显示模式显示的track列表
2.track行
name=<track_label> 定义track的标签
description=<center_label> 定义显示的时候track的中间的标签
type=<track_type> 定义track类型,可以定义为BAM, BED detail, bedGraph, bigBed, bigWig, broadPeak, narrowPeak, Microarray, VCF and WIG
visibility=<display_mode> 定义显示模式,定义track的起始显示模式,包括0 - hide, 1 - dense, 2 - full, 3 - pack, and 4 - squish
color=<RRR,GGG,BBB> 定义注释track的主演色,包括三个逗号分隔的0-255之间的数字,默认0,0,0黑色
itemRgb=On 如果开了这个选项,bed文件定义的itemRgb生效
colorByStrand=<RRR,GGG,BBB,RRR,GGG,BBB> 设置正负链的颜色,默认0,0,0,0,0,0 都是黑色
useScore=<use_score> 默认是0,使用bed score值定义的颜色,如果是1,会使用数据行来决定颜色深浅
group=<group> 定义track组,会在genome browser上显示
priority=<priority> 定义组内排列位置,没有分组的话会定义默认组(user)的排列位置
db=<UCSC_assembly_name> 定义要比对的数据库,比如hg18,mm8等
offset=<offset> 补偿,定义添加到全部坐标上的数值,默认0
maxitems<#> 定义track能包括的最大条目,默认250,必须小心设置,不然会导致系统不稳定
url=<external_url> 定义track 的额外链接内容
htmlUrl=<external_url> 定义track描述页面的链接内容
bigDataUrl=<external_url> 定义数据文件的url,就是放在服务器上的文件地址,
下面是UCSC给出的例子
1 browser position chr21:33,031,597-33,041,570 2 track type=bigBed name="bigBed Example One" description="A bigBed file" bigDataUrl=http://genome.ucsc.edu/goldenPath/help/examples/bigBedExample.bb
bed文件格式可以直接写入track中,如下,在基因组固定位置显示蓝色和绿色标记
1 browser position chr22:20100000-20140000 2 track name=spacer description="Blue ticks every 10000 bases" color=0,0,255, 3 chr22 20100000 20100001 4 chr22 20110000 20110001 5 chr22 20120000 20120001 6 track name=even description="Red ticks every 100 bases, skip 100" color=255,0,0 7 chr22 20100000 20100100 first 8 chr22 20100200 20100300 second 9 chr22 20100400 20100500 third
2.2 UCSC 的MyData下的track hub
track hub 是track 的收集,hub中的track在genome browser浏览页面中以蓝色显示。
首先在存放hub文件的文件夹下写一个hub文件,格式如下
1 hub hub_name # hub的名称 2 shortLabel hub_short_label #hub的短标签,便于显示 3 longLabel hub_long_label #hub具体的标签 4 genomesFile genomes_filelist #要对比的基因组列表文件路径 5 email email_address #自己的email地址 6 descriptionUrl descriptionUrl # 对这个track的描述
接下来编辑基因组列表文件
1 genome assembly_database_1 #对比到的基因组,比如hg19 2 trackDb assembly_1_path/trackDb.txt #trackDb文件,包括对比到hg19的所有track 3 4 5 genome assembly_database_2 6 trackDb assembly_2_path/trackDb.txt
上面是两个不同的trackDb(track数据库),分别对比到不同的基因组,而trackDb中写入有很多不同的对比到该基因组的hub track
最后编辑trackDb文件
1 track dnaseSignal #在genome browser上显示的track名,必须独一无二 2 bigDataUrl dnaseSignal.bigWig #文件的url,默认在trackDb所在文件夹 3 shortLabel DNAse Signal 4 longLabel Depth of alignments of DNAse reads 5 type bigWig 6 7 8 track dnaseReads 9 bigDataUrl dnaseReads.bam 10 shortLabel DNAse Reads 11 longLabel DNAse reads mapped with MAQ 12 type bam
上面写入了两个track,一个是bigWig格式的文件,一个是bam文件.而vcf 文件如下
1 track GC_WGS_tumour_vcf_by_lumpy 2 type vcfTabix 3 bigDataUrl GC_WGS_tumour.sorted.vcf.gz 4 shortLabel GC_WGS tumour vcf lumpy 5 longLabel GC_WGS tumour vcf by lumpy
上面几个是基本参数,更多可选的hub track的参数参见hub track 定义文档
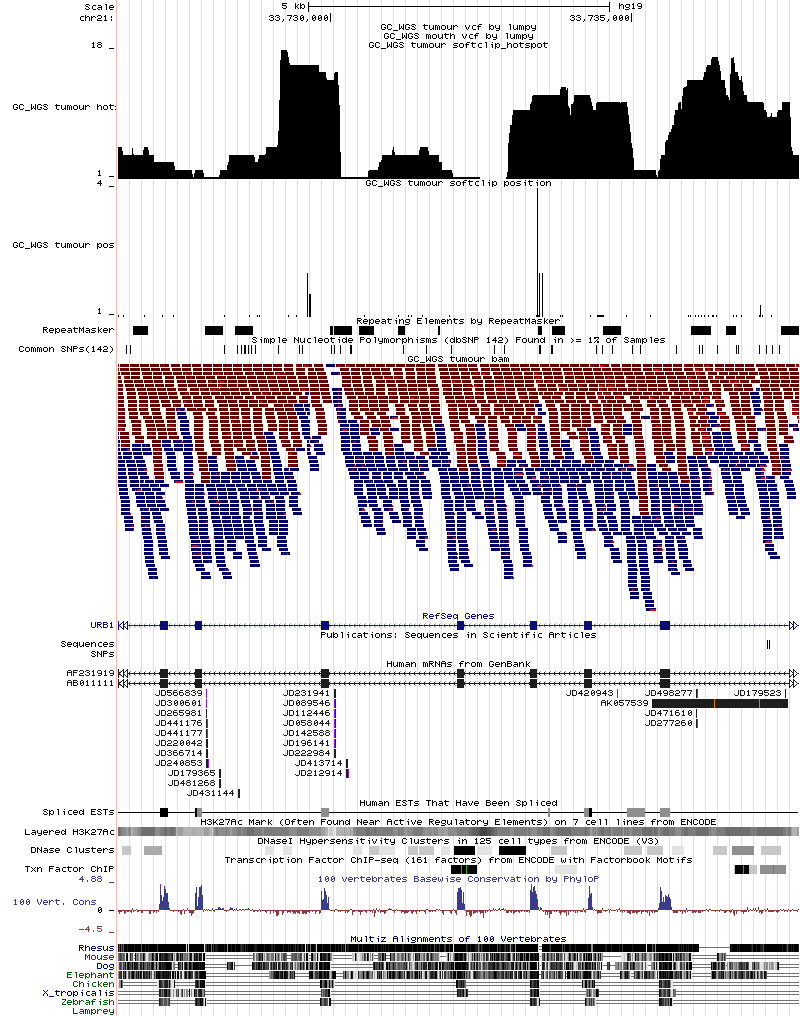
最后上图一张
参考文献
UCSC custom track: http://genome-asia.ucsc.edu/goldenPath/help/customTrack.html
UCSC track hub : http://genome-asia.ucsc.edu/goldenPath/help/hgTrackHubHelp.html
UCSC hub track 定义文档 :http://genome-asia.ucsc.edu/goldenPath/help/trackDb/trackDbHub.html