DiffBind是基于peak的差异分析包,peaks由其他peak caller软件生成,如MACS2、HOMER.一个peak可能表示一个染色质开放区域、蛋白质结合位点等。call出来的peak是包含它的染色体、开始和结束位置信息的,然后可以通过bam文件根绝位置信息获取在peak上的read数量。进一步通过DESeq2或edgeR进行核心的差异分析。
官方文档:http://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf
本文是根据官方文档所写的一点笔记。详细内容请移步官网文档。
DiffBind安装
在R里使用以下命令安装
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("DiffBind")
要是报错的话,就去搜一下报错信息,可能R版本不对,或者没权限等问题。
基本使用
初始输入信息
DiffBind输入的文件需要一个sample sheet,可以从预先填写好的csv文件中读取,或者构建一个dataframe。
完整的字段包括这些
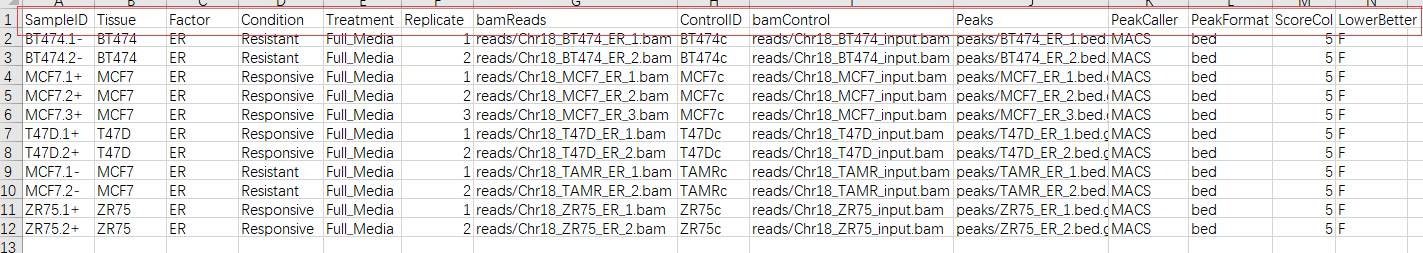
不过构建csv文件时,也不需要全部都填上。SampleID,bamReads(bam文件地址),Peaks(peak文件地址)填好,其他的根据实际情况填写或者默认就好。
在ATAC-Seq数据中没有control,controlID和bamControl删去就好。PeakCaller和PeakFormat用一个就好,支持的格式有

下面以这个sample.test.csv为例做演示:
1 SampleID,Factor,Replicate,bamReads,Peaks,PeakCaller,Tissue
2 tt.e11.5.Rep1,tt.e11.5,1,/home/user/project/atac_seq/bam/tt.e11.5.rep1.bt2.mm10.sort.filter.shift.bam,/home/user/project/atac_seq/callPeak/tt.e11.5.rep1_peaks.narrowPeak,narrow,tt
3 tt.e11.5.Rep2,tt.e11.5,2,/home/user/project/atac_seq/bam/tt.e11.5.rep2.bt2.mm10.sort.filter.shift.bam,/home/user/project/atac_seq/callPeak/tt.e11.5.rep2_peaks.narrowPeak,narrow,tt
4 tt.e15.5.Rep1,tt.e15.5,1,/home/user/project/atac_seq/bam/tt.e15.5.rep1.bt2.mm10.sort.filter.shift.bam,/home/user/project/atac_seq/callPeak/tt.e15.5.rep1_peaks.narrowPeak,narrow,tt
5 tt.e15.5.Rep2,tt.e15.5,2,/home/user/project/atac_seq/bam/tt.e15.5.rep2.bt2.mm10.sort.filter.shift.bam,/home/user/project/atac_seq/callPeak/tt.e15.5.rep2_peaks.narrowPeak,narrow,tt
Reading in the peaksets:
> dbObj <- dba(sampleSheet="./sample.test.csv")
Counting reads:
> dbObj <- dba.count(dbObj , bUseSummarizeOverlaps=TRUE)
额,作者说参数bUseSummarizeOverlaps用更加标准的方法来counting。
bUseSummarizeOverlaps
logical indicating that summarizeOverlaps should be used for counting instead of the built-in counting code.
This option is slower but uses the more standard counting function. If TRUE, all read files must be BAM (.bam extension),
with associated index files (.bam.bai extension). The insertLength parameter must absent.
See notes for when the bRemoveDuplicates parameter is set to TRUE, where the built-in counting code
may not correctly handle certain cases and bUseSummarizeOverlaps should be set to TRUE.
Establishing a contrast:
这是建立一个分组,指定哪些样本是一组的。
可以使用参数categories自动构建分组,DBA_FACTOR是DiffBind中定义的常量,类似的常量都是以"DBA_"开头,如DBA_ID, DBA_TISSUE,DBA_CONDITION
minMembers是每组最小成员数目。
## 将根据factor分为两组
> dbObj <- dba.contrast(dbObj, categories=DBA_FACTOR, minMembers = 2)
> dbObj
4 Samples, 28477 sites in matrix:
ID Tissue Factor Replicate Caller Intervals FRiP
1 tt.e11.5.Rep1 tt tt.e11.5 1 counts 28477 0.11
2 tt.e11.5.Rep2 tt tt.e11.5 2 counts 28477 0.12
3 tt.e15.5.Rep1 tt tt.e15.5 1 counts 28477 0.19
4 tt.e15.5.Rep2 tt tt.e15.5 2 counts 28477 0.12
1 Contrast:
Group1 Members1 Group2 Members2
1 tt.e11.5 2 tt.e15.5 2
Performing the differential analysis
dbObj <- dba.analyze(dbObj, method = DBA_DESEQ2)
默认使用DESeq2进行计算,可以选择DBA_EDGER(edgR),或者两个都要DBA_ALL_METHODS
韦恩图绘制
> dba.plotVenn(dbObj, mask=dbObj$masks$nt)
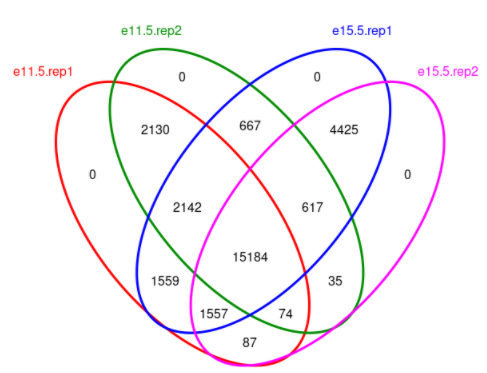
mask是用于选择哪些样本用于绘图。
## 内置的一些mask
> dbObj$masks
$nt
nt.e11.5.Rep1 nt.e11.5.Rep2 nt.e15.5.Rep1 nt.e15.5.Rep2
TRUE TRUE TRUE TRUE
$nt.e11.5
nt.e11.5.Rep1 nt.e11.5.Rep2 nt.e15.5.Rep1 nt.e15.5.Rep2
TRUE TRUE FALSE FALSE
$nt.e15.5
nt.e11.5.Rep1 nt.e11.5.Rep2 nt.e15.5.Rep1 nt.e15.5.Rep2
FALSE FALSE TRUE TRUE
......
> dba.plotVenn(dbObj, mask=dbObj$masks$Replicate.2)

保存结果
write.csv(dbObj.report, file="~/tmp/test.csv")